Download

1 / 12
250 likes | 536 Vues
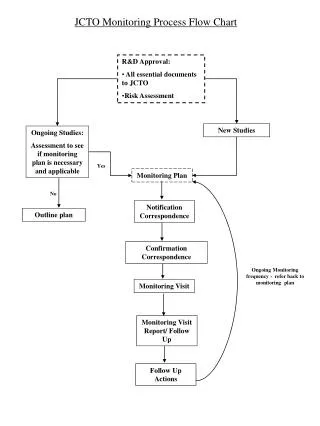
Module H Interim Monitoring Visit. Denise Thwing 21 Apr 2010. Purpose Interim Monitoring Visit. Rights and well being of subjects are protected New information regarding drug is shared Verify site compliance with the protocol Confirm data points SDV Verify site regulatory adherence

E N D
Module H Interim Monitoring Visit Version: Final 21-Apr-2010 Denise Thwing 21 Apr 2010
Purpose Interim Monitoring Visit • Rights and well being of subjects are protected • New information regarding drug is shared • Verify site compliance with the protocol • Confirm data points • SDV • Verify site regulatory adherence • Approved ICF • Prompt reporting AEs/deviations Version: Final 21-Apr-2010
Purpose Interim Monitoring Visit • Objectives • Scientific • Verify clinical results represent a valid assessment of the drug’s effectiveness and safety. • QOL assessments • Improvement in baseline assessments • Stable /expected change VS • AEs/SAEs/drug related? • Lab results close to baseline • Regulatory • Make sure conditions are met for the acceptance of the data by appropriate regulatory authorities • ICFs review/multiple versions • Report AEs/deviations • GCP requirements • Managerial/economic • Make sure the first two objectives are reached in a timely and effective manner • Study conducted per schedule • Queries resolved promptly and accurately • Site/subject compensation Version: Final 21-Apr-2010
Philosophy • Many problems can trace back to a lack of, or unclear communication • Monitoring a study takes a lot of thought and preparation upfront • Sites with little or no enrollment may lack enthusiasm • CRA/sponsor presence indicates to a site they are important or under scrutiny • Booster visits Version: Final 21-Apr-2010
Preparing for the visit • MV interval specified in SOW and or SOPs • First visit typically within 2 weeks of FPI • Complexity of protocol • Phase III study with many different inclusion /exclusion criteria • Disease being evaluated • Infectious disease or HTN study • Phase of study • CRF pages > 60 days? • DCFs > 5 business days? • CRC may need assistance answering DCFs • Previous site performance • Enrolled high number subjects • CRA experience and effectiveness Version: Final 21-Apr-2010
Preparing for the visit • Confirmation letter • Site visit log • SOPs • IMV template/MV checklist • Cross check data points • Subject drug accountability log matches master inventory log • Regulatory document review checklist • CMP • Review outstanding action items Version: Final 21-Apr-2010
Conducting the visit • Record number screened, enrolled, screen failed and dropped/completed • Enrollment log • Do all meet inclusion/exclusion criteria? • Review all ICFs • Confirm versions • New SAEs/reported? • End ongoing AEs/meds • Review any staff changes/facility changes • Is replacement qualified by experience, education and protocol training performed? • Has CV been obtained & sponsor notified? • Impacts any change to regulatory documents? • All tasks performed as per delegation log? Version: Final 21-Apr-2010
Conducting the visit • Source data verification • 100% review vs. key safety and efficacy variables • Legible • Meets coding requirements • Left sided nodule • Magic mouthwash • New Concomitant? • Questionable data? • Record which visits have been reviewed and which CRFs collected • Needed for analysis • Drives payment process Version: Final 21-Apr-2010
Conducting the visit • Drug accountability • Correct treatment group • Subject compliance • Study supplies adequate? • Lab procedures as per protocol • Supplies adequate • Review regulatory binder • Advertising approvals • IND safety reports reported • Deviations reported • Review outstanding action items • Record new action items • Review ISF • Meet with PI and CRC • Follow-up letter Version: Final 21-Apr-2010
Adverse Events • A serious adverse event (SAE) in human drug trials are defined as any untoward medical occurrence that at any dose • results in death – direct outcome of the adverse event e.g. device implant • is life-threatening – suspected use product may result in death e.g. pacemaker failure • requires inpatient hospitalization or prolongation of existing hospitalization, e.g. allergic reaction, bleeding • results in persistent or significant disability/incapacity e.g. CVA due to drug induced hypercoagulability • is a congenital anomaly/birth defect e.g. thalidomide • Requires intervention to prevent permanent disability – burns from radiation equipment The term "life-threatening" in the definition of "serious" refers to an event in which the patient was at risk of death at the time of the event; it does not refer to an event which hypothetically might have caused death if it were more severe. Adverse events are further defined as “Any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product and which does not necessarily have to have a causal relationship with this treatment.” Wikipedia Version: Final 21-Apr-2010
Top Five deficiencies - FDA Audits • Failure to follow protocol • Failure to keep adequate records • Problems with the ICF • Failure to report adverse events • Failure to account for the disposition of the study drug Version: Final 21-Apr-2010
Questions? Version: Final 21-Apr-2010