Download
1 / 37
380 likes | 573 Vues
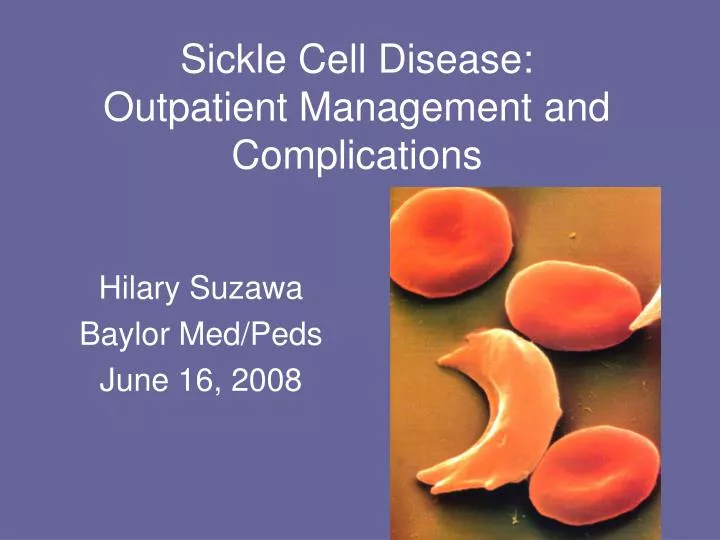
Sickle Cell Disease: Outpatient Management and Complications. Hilary Suzawa Baylor Med/Peds June 16, 2008. Sickle Cell Disease. First described 1910 Ethnic backgrounds: Black (African) Mediterranean Arabian peninsula Indian subcontinent Caribbean South and Central America.

E N D
Sickle Cell Disease: Outpatient Management and Complications Hilary Suzawa Baylor Med/Peds June 16, 2008
Sickle Cell Disease • First described 1910 • Ethnic backgrounds: • Black (African) • Mediterranean • Arabian peninsula • Indian subcontinent • Caribbean • South and Central America
Hemoglobin S • Substitution of a valine for glutamic acid at the sixth amino acid of the beta globin chain • Produces a hemoglobin tetramer (alpha2/beta S2) that is poorly soluble when deoxygenated
Variants • Hemoglobin SS (SCD-SS) • Hemoglobin SC (SCD-SC) • Sickle Beta Thalassemia • SCD-SB+thal or SCD-SB0 thal • In US ~2/3 of SCD are SCD-SS • SCD-SC or Sickle Beta Thalassemia may NOT have significant anemia
Average Lifespan • Sickle Cell Disease (SCD-SS)— • 45 years • One generation ago average survival was as low as 14 years • SCD-SC or Sickle Beta Thalassemia— • 50-60 years
Prenatal or Preconception • Amniocentesis if needed • Genetics evaluation • High-risk ob if needed • Maternal mortality <2% and neonatal mortality <5% • Increased risk of pre-eclampsia, pre-term labor, low-birth weight
Birth-6 months • Visit Q2 months (2 wk, 2 mth, 4 mth, 6 mth) • CBC every visit • Heptavalent conjugated pneumococcal vaccine (Prevnar) at 2 mth, 4 mth, 6 mth • PCN VK 125 mg po BID starting at 2-3 mths
6 months-2 years • Visit Q3 months (6 mth, 9 mth, 12 mth, 15 mth, 18 mth, 21 mth, 2 yrs) • CBC every 3-6 mths • UA annually • Ferritin or serum iron and TIBC once at 1-2 yrs • BUN, Cr, LFTS once at age 1-2 yrs • Prevnar at 15 mths • Flu vaccine • Start folic acid daily at 1 year • Consultation with pedi heme
2 years • Screening with specialized Doppler US transcranial blood-flow velocity (TBV) • Recommended for asymptomatic children with sickle cell dz beginning at age 2 yrs • Recommended annually from 2-16 yrs age • TBV 200 cm/sec or more are at increased risk of stroke
2 years-5 years • Visit Q6 months • CBC at least yearly • UA at least yearly • BUN, Cr and LFTs every 1-2 yrs • 23-valent Pneumococcal vaccine (Pneumovax) at age 2 yrs; booster at age 5 yrs • Meningococcal vaccine after age 2 yrs • Flu vaccine annually • Increase PCN VK to 250 mg po BID at 3 yrs old
>5 years • Visits Q6-12 months • CBC at least yearly • UA at least yearly • BUN, Cr and LFTs every 2-3 years • Flu vaccine yearly • Consider stop PCN VK • NOT if pt has had splenectomy OR h/o invasive pneumococcal infxn • Continue folic acid • Annual eye exam starting at 10 yrs (focus on retina)
Growth • Decrease in height and weight compared to peers • During adolescent growth spurt begin to catch up with peers • Do NOT experience growth failure, only growth delay • Delayed puberty • Menarche delayed 2-2.5 years • Dysmenorrhea may precipitate pain episodes • Puberty, although delayed, proceeds normally once started
Adolescence • Visits annually • CBC yearly • UA yearly • BUN, Cr and LFTs every 2-3 years • Ferritin or serum iron and TIBC at least once • Flu vaccine yearly • Transition from pediatric to adult care
Adult • Biannual visit for pt >30 years • Annual eye exam • Vaccinations • Hepatitis vaccine • Pneumococcal vaccine (Pneumovax) • Evaluate for elevated BP and pulmonary HTN • Foot care and protective shoes • Consider PCN VK prophylaxis • Folic acid daily to ensure adequate amounts for high erythrocyte turnover • Consider hydroxyurea
Pneumovax • First dose at 2 years • If pt <10 years, then give booster at age 3-5 years • If pt >10 years, then give booster 5 years after first dose • Support for a one time booster 5 years after first dose; some institutions give Q5 years
Blood Pressure • Pt with SCD-SS have lower systolic and diastolic blood pressure compared to others • Modest increases in BP within the normal range for general population may indicate underlying renal dz
Pulmonary Hypertension • Monitor for pulmonary hypertension • Unexplained dyspnea or hypoxemia • Loud P2 • Enlarged heart (RV) on CXR • Check Echo
PCN VK prophylaxis • By age 2 months, start PCN VK 125 mg po BID • Age 3 years increase dose to PCN VK 250 mg po BID • Functional asplenia occurs <1 year age in pt with SCD-SS and SCD SB0thal • SCD-SS patients have usually infarcted their spleens before 4 years • Can be discontinued after age 5 years except in child who has had splenectomy or invasive infxn
Hydroxyurea • Increases Hb F levels • Improves red cell deformability and hydration • Reduces red cell adhesion to vascular endothelium
Blood Transfusion • Post-transfusion, Hb level should not exceed 10-11 g/dL • Check for infection: HIV, Hepatitis panel • Alloimmunization—in up to 30% of pt who receive frequent transfusions • Volume overload • Iron overload • Use of chelator • Exchange transfusion
Clinical Case #1 • 2 yo AAM with h/o SCD-SS presents for sick visit. • On physical exam pt has palpable spleen not previously observed. • Pt’s baseline Hb ~8 but current Hb ~3 • Reticulocyte count increased from pt’s baseline
Clinical Case #1 • What medical condition do you suspect? • Acute Splenic Sequestration Crisis: sudden enlargement of spleen, precipitous fall in Hct (at least 2 g/DL), rise in reticulocyte count. • What is most common age for this to develop? • Often occurs between 6 months and 3 years • Hb SS at age <3 years. Variants at any time during childhood.
Clinical Case #1 • What treatment do you recommend? • Hospitalization • IVF +- blood transfusion • After first or second crisis, consider elective splenectomy when child is stable • Preventive education: Caretaker should know how to palpate for splenic size and know about risk of sequestration crisis
Clinical Case #2 • 17 yo AAF with h/o RAD and SCD-SS was recently hospitalized for a vaso-occlusive pain crisis. Presents 1 week after discharge for hospital follow-up. • Now c/o minimal bilateral LE pain, chest discomfort, fever (T=100) • Pulse ox in the office Sat 88% on RA • Chest X-ray: Normal
Clinical Case #2 • What medical condition do you suspect? • Acute Chest Syndrome • Ages birth-9 years • May be accompanied by or preceded by pain the chest or extremities, fever, respiratory distress and low O2 Sat • Hypoxemia is not necessary for dx • CXR may be normal initially with later development of infiltrate • 30% of all Hb SS pt will have one episode of Acute Chest Syndrome; ½ of these pt will have recurrent episodes • Asthma increases risk of ACS
Clinical Case #3 • 5 yo AAM with SCD-SS disease presents with a new limp • What medical conditions would you consider? • What other information would you like to know?
DDX Limp in Hb SS Disease • DDX • Osteomyelitis • Avascular Necrosis of the hip • Stroke • Other information: • H/o HA • Fever • Labs: WBC, ESR • Extremity exam, hip exam • Neuro exam
AVN • Most common site of AVN • Femoral head • Humeral head • Knee • Small joints of hands and feet • Most common orthopedic complication in SCD and occurs in up to 50% of all pt • SCD is the most common cause of AVN in children
Stroke • Stroke occurs in 7-11% of children with SCD • SCD-SS overall risk of stroke 3.75% throughout lifetime • <2 yrs have lowest incidence of stroke • 2-5 years have highest incidence infarctive stroke • 20-29 years have highest incidence hemorrhagic stroke
Questions • What is the most common cause of osteomyelitis in pt with SCD-SS? • Salmonella #1, S. aureus #2 • How much does Hb usually drop with aplastic crisis? • >= 30% reduction from baseline Hb • What is the most common cause of aplastic crisis? • Parvovirus B19 causes ~70%
Bacterial infection • What are most common bacteria causing infxn in SCD-SS? • Streptococcus pneumoniae • Staphylococcus aureus • Salmonella species • Haemophilus influenza type b • Escherichia coli • Klebsiella species
Complications • Airway/Breathing • Acute chest syndrome • Chronic Pulmonary Hypertension • CNS • Stroke • Pain • Cardiovascular • Hypertension
GI Complications • GI • Cholelithiasis, Choledocholithiasis, Acute Cholecystitis • Abdominal Pain Crisis • Sickle Hepatopathy • Pancreatitis • Peptic Ulcer Disease • Hematologic • Acute splenic sequestration crisis • Aplastic crisis • Blood transfusion complications • GU/Gyn • Priapism • Pregnancy Complications, need for Genetic Counseling
Complications • Infection • Osteomyelitis • Renal Disease • Orthopedic • Aseptic Necrosis of the hip (AVN) • Chronic Leg Ulcers • Multiple Organ Failure Syndrome: • fever, decrease Hb, decrease plt, rhabdomyolysis, mental status changes
Bibliography • Koshy, M and Dorn, L. “Continuing Care for Adult Patients with Sickle Cell Disease.” Hematology/Oncology Clinics of North America 1996; 10 (6): 1265-1273. • Lottenberg, R and Hassell, K. “An Evidence-Based Approach to the Treatment of Adults with Sickle Cell Disease.” Hematology 2005; 58-65. • Mehta, S et al. “Opportunities to Improve Outcomes in Sickle Cell Disease.” American Family Physician 2006; 74 (2): 303-310.
Bibliography • Redding-Lallinger, R and Knoll, C. “Sickle Cell Disease—Pathophysiology and Treatment.” Curr Probl Pediatr Adolesc Health Care 2006; 36: 346-376. • Wethers, D. “Sickle Cell Disease in Childhood: Part I. Laboratory Diagnosis, Pathophysiology and Health Maintenance.” American Family Physician 2000; 62 (5): 1013-1020.
Bibliography • Wethers, D. “Sickle Cell Disease in Childhood: Part II. Diagnosis and Treatment of Major Complications and Recent Advances in Treatment.” American Family Physician 2000; 62 (6): 1309-1314.