Download
1 / 1
10 likes | 111 Vues
Table 1. Pompe’s Disease : Amino Acid Changes and Effects. Joel Bucci , Jennifer Ryan, and Dylan Storey.

E N D
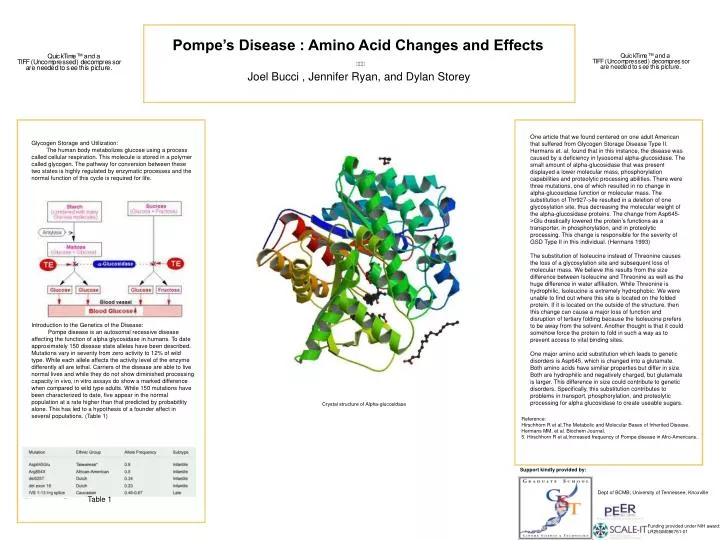
Table 1 Pompe’s Disease : Amino Acid Changes and Effects Joel Bucci , Jennifer Ryan, and Dylan Storey One article that we found centered on one adult American that suffered from Glycogen Storage Disease Type II. Hermans et. al. found that in this instance, the disease was caused by a deficiency in lysosomal alpha-glucosidase. The small amount of alpha-glucosidase that was present displayed a lower molecular mass, phosphorylation capabilities and proteolytic processing abilities. There were three mutations, one of which resulted in no change in alpha-glucosidase function or molecular mass. The substitution of Thr927->Ile resulted in a deletion of one glycosylation site, thus decreasing the molecular weight of the alpha-glucosidase proteins. The change from Asp645->Glu drastically lowered the protein’s functions as a transporter, in phosphorylation, and in proteolytic processing. This change is responsible for the severity of GSD Type II in this individual. (Hermans 1993) The substitution of Isoleucine instead of Threonine causes the loss of a glycosylation site and subsequent loss of molecular mass. We believe this results from the size difference between Isoleucine and Threonine as well as the huge difference in water affiliation. While Threonine is hydrophilic, Isoleucine is extremely hydrophobic. We were unable to find out where this site is located on the folded protein. If it is located on the outside of the structure, then this change can cause a major loss of function and disruption of tertiary folding because the Isoleucine prefers to be away from the solvent. Another thought is that it could somehow force the protein to fold in such a way as to prevent access to vital binding sites. One major amino acid substitution which leads to genetic disorders is Asp645, which is changed into a glutamate. Both amino acids have similiar properties but differ in size. Both are hydrophilic and negatively charged, but glutamate is larger. This difference in size could contribute to genetic disorders. Specifically, this substitution contributes to problems in transport, phosphorylation, and proteolytic processing for alpha glucosidase to create useable sugars. Glycogen Storage and Utilization: The human body metabolizes glucose using a process called cellular respiration. This molecule is stored in a polymer called glycogen. The pathway for conversion between these two states is highly regulated by enzymatic processes and the normal function of this cycle is required for life. Introduction to the Genetics of the Disease: Pompe disease is an autosomal recessive disease affecting the function of alpha glycosidase in humans. To date approximately 150 disease state alleles have been described. Mutations vary in severity from zero activity to 12% of wild type. While each allele affects the activity level of the enzyme differently all are lethal. Carriers of the disease are able to live normal lives and while they do not show diminished processing capacity in vivo, in vitro assays do show a marked difference when compared to wild type adults. While 150 mutations have been characterized to date, five appear in the normal population at a rate higher than that predicted by probabitlity alone. This has led to a hypothesis of a founder affect in several populations. (Table 1) Crystal structure of Alpha-glucosidase Reference: Hirschhorn R et al,The Metabolic and Molecular Bases of Inherited Disease. Hermans MM. et al. Biochem Journal. 5. Hirschhorn R et al.Increased frequency of Pompe disease in Afro-Americans.. Support kindly provided by: Dept of BCMB; University of Tennessee, Knoxville Funding provided under NIH award: LR25GM086761-01