Download
1 / 48
480 likes | 806 Vues
The Role of the Registered Dietitian in the Nutritional Management of Cystic Fibrosis Disease Progression. Hayley Kurtz Sodexo Dietetic Internship March 6, 2014. Agenda. Cystic Fibrosis CFTR Gene & Organs Diagnosis Symptoms & Complications Medical Treatment Medical Nutrition Therapy

E N D
The Role of the Registered Dietitian in the Nutritional Management of Cystic Fibrosis Disease Progression Hayley Kurtz Sodexo Dietetic Internship March 6, 2014
Agenda • Cystic Fibrosis • CFTR Gene & Organs • Diagnosis • Symptoms & Complications • Medical Treatment • Medical Nutrition Therapy • Presentation of Patient • Critical Comments • Summary
CFTR Gene • Cystic fibrosis transmembrane conductance regulator (CFTR) gene • CF related to defects in CFTR gene makeup • CFTR gene produces CFTR protein • Made up of 1,480 amino acids • Each domain acts as a channel transporting chloride across cell membranes
Classifications of Mutations According to Their Effect on the CFTR Protein • Class I, II and III mutations generally lead to complete loss of function and a more severe disease. • Class IV and V cause a reduction in function and have a milder effect.
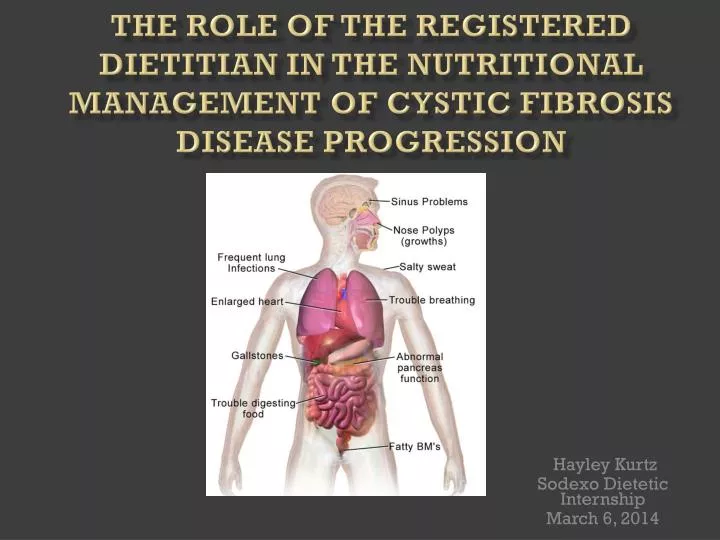
CFTR Gene Mutations Affect: • Sinus • Pancreas • Lungs • Sweat glands • Reproductive tract
Pancreas • Exocrine and endocrine cells • CFTR’s role
Pancreas • CFTR protein defect: absence or dysfunction of CFTR channel in pancreatic ductules
Lungs • Expands and contracts ~20 times per minute to supply oxygen to tissues • Functions of CFTRproteinchannel: • Transports chloride in and out of cells • Transports sodium in and out of the airway surface liquid • Bicarbonate neutralizes chloride ions
Lungs Healthy Lungs CF Lungs
Diagnosis of Cystic Fibrosis • Newborns inherit two copies of the CFTR gene, one from each parent
Diagnostic Testing • Prenatal testing • Sweat-chloride test • Chest x-ray • Sinus x-ray • Lung function test • Sputum culture
Complications • Buildup of thick, sticky mucus • Excessively salty skin • Pancreatic insufficiency • Malnutrition • Chronic lung infections • Bronchiectasis • Pneumothorax
Medical Treatment Thoracic Expansion Exercises Forced Expiration Technique Autogenic Drainage Percussion Vibrations Huffing
Medical Nutrition Therapy • Multidisciplinary team • Pulmonologist/Pediatric Pulmonologist • Registered Dietitian • Social Worker • Critical Care Specialist • Pulmonary Surgeon • Respiratory Care Practitioner • Registered Nurse • Primary Care Physician • Endocrinologist, Gastroenterologist & Cardiologist
Medical Nutrition Therapy • Early detection • Monitor growth and nutritional status • Lung function and malnutrition interrelated
Nutrition Assessment • Indicators for nutritional status: • Genetic potential • Growth charts • Weight • Puberty development
Pancreatic Enzyme Replacement • Starting dose: • 500 units/kg/meal • 250 units/kg/snack • Increasing gradually until 10,00 units/kg/day is reached • Do not exceed 10,000 units/kg/day
Laboratory Data • Blood glucose • Arterial blood gases • Lipase • Amylase • Sodium • Chloride
Medical & Social History of R.L. • Age: 26 • Sex: Female • Race: Caucasian • Allergies: NKFA • Height: 63in (160cm) • Weight: 81.9 lbs (37kg) • BMI: 14.5 • PMH: CF, Dehydration, Malnutrition, Depression, Bronchiectasis, Pulmonary HTN • Nutrition support: PEG tube receiving nocturnal feeds of enteral nutrition (Perative) & bolus feeds PRN at home • Family hx: Brother passed away from CF • Treated at Penn-Presbyterian for the last 7 years by a Pediatrician who specializes in CF
PPMC Admission • November 11, 2013: Admission • Dx: CF exacerbation, 9% wt loss x 1 month, dehydration, malnutrition, change in cough sputum, FEV1 12-13% • November 12, 2013: R.L. declared as VIP • Initial assessment
Day OneAssessment • 81.9 lbs/37kg • 63in/160cm • BMI: 14.5 • %IBW: 70% • %UBW: 91% • 9% wt loss x 1 month • Abnormal labs: Glucose 134mg/dL, BUN 6mg/dL, Magnesium 1.6mg/dL • Medications: Vit D, Vibramycin, Iron Sulfate, Prevacid, Reglan, MVI, Pancrealipase
Day OneNutrition Requirements • Increased needs per CF, intubated and extubated needs dependent of medical status: • Intubated: 1100-1290kcals/day (30-32kcals/kg) • Extubated: 1480-1850kcals/day (40-50kcals/kg) • Protein: 56-74g/day (1.5-2.0g/kg) • Fluid: 1100-1290mL/day (30-32mL/kg)
Day OneNutrition Prescription • Intubated: Perative @ 40mL/hr x 24 hrs + bolus flushes 60mL q6hrs to provide 1248kcals, 64g protein, 1120mL • Extubatedw/ bipapx 12 hrs: Perative @ 100mL x 12 hrs + bolus flush 200mL x 1 daily to provide 1560kcals, 80g protein, 1150mL • Extubatedw/ bipap >12 hrs: Recommend post-pyloric feeds (J tube) Perative @ 55mL/hr x 24 hrs + bolus flush 110mL x 1 daily to provide 1716kcals, 88g protein, 1150mL
Day TwoFollow up • Remains on bipap • Medical staff requesting TPN recommendations 2/2 patient requiring bipap >12 hrs • Medical staff did not take my recommendation of post pyloric feeds via J-tube while on bipap >12 hrs • Spoke with medical staff to replete electrolytes prior to initiating TPN • Identified patient with severe weight loss may cause risk of refeeding
Day TwoNutrition Intervention • TPN recommendations • Initiate TPN day 1: 340kcals Dextrose, 54 g AA • Continue to monitor electrolytes, if depleted, replete prior to advancing TPN (waiting 24-48 hrs of electrolytes WNL) • TPN day 2: 340kcals Dextrose, 75g AA, 166kcals Lipids • TPN day 3: 510kcals Dextrose, 75 g AA, 246kcals Lipids • Goal: 544kcals Dextrose, 75g AA, 370kcals Lipids • 1214 kcals, 914 NPC, GIR 3 meeting 82% estimated kcal needs, 101% estimated protein needs
Day FourFollow up • Remains on bipapx 24 hrs, NPO • Electrolytes WNL • TPN initiated • Day 1 TPN recommendations
Day 8Follow up • Remains on bipapx 24 hrs, NPO • Labs not being drawn • TPN has not advanced • Stage I pressure ulcer on nose r/tbipap • Full code changed to DNAR-B
Day 10Follow up • Remains on bipapx 24 hrs • Rapid pulmonary function deterioration • TPN still not advanced • R.L. passed away in the MICU this evening
Critical Comments • Stable at previous admissions • Appropriate clinical judgment • Noncompliant with TF and PO dietary intake • Weight loss during initial assessment • Medical team decided against nutrition recommendations
References • U.S. National Library of Medicine. Genetics Home Reference: CFTR. National Institute of Health.http://ghr.nlm.nih.gov/gene/CFTR. Published January 6, 2014. Accessed January 8, 2014. • Cystic Fibrosis Foundation. Living with Cystic Fibrosis. http://www.cff.org/treatments/. Accessed December 22, 2013. • Brown, MB, Haack, KKV, Pollack, BP, et al. Low abundance of sweat duct Cl− channel CFTR in both healthy and cystic fibrosis athletes with exceptionally salty sweat during exercise. American Journal of Physiology. http://ajpregu.physiology.org/content/300/3/R605. 2011; 30: R605-R615. DOI: 10.1152/ajpregu.00660.2010. • National Heart, Lung, and Blood Institute. How is cystic fibrosis diagnosed? National Institute of Health.http://www.nhlbi.nih.gov/health/health-topics/topics/cf/diagnosis.html. Published December 26, 2013. Accessed December 29, 2013. • US National Library of Medicine. The role of cftr in bicarbonate secretion by pancreatic duct and airway epithelia. National Institute of Health. http://www.ncbi.nlm.nih.gov/pubmed/20224219. Published 2009. Accessed December 29, 2013. • Mayo Clinic Staff. Cystic fibrosis: disease complications. Mayo Clinic. http://www.mayoclinic.org/diseases-conditions/cystic-fibrosis/basics/complications/CON-20013731. Published June 13, 2012. Accessed December 22, 2013. • American Lung Association. Cystic fibrosis: state of lung disease in diverse communities. http://www.lung.org/assets/documents/publications/solddc-chapters/cf.pdf. Published 2010. Accessed January 7, 2014.
References • Lucile Packard Children’s Hospital Pediatric Pulmonogists Team. Pulmonary medicine and cystic fibrosis. Stanford Children’s Health. http://www.lpch.org/clinicalSpecialtiesServices/COE/PulmonaryCareCF/team.html. Accessed January 4, 2014. • Accurso FJ, Castellani C, Cutting GR, et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: cystic fibrosis foundation consensus report. J Pediatrics. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2810958/pdf/nihms125156.pdf. 2008; 153(2): S4-S14. DOI: 10.1016/j.jpeds.2008.05.005. • America Thoracic Society. To sweat or not to sweat: is that enough to diagnose cystic fibrosis? American Journal of Respiratory and Critical Care Medicine.http://www.atsjournals.org/doi/pdf/10.1164/rccm.201208-1510EDD. 2012; 186: 700-701. DOI: 10.1164/rccm.201208-1510ED. • Stanford University Psychology Department. Human genome, cystic fibrosis. Stanford University. http://www.stanford.edu/class/psych121/humangenome-CF.htm#sweat. Published 2010. Accessed December 22, 2013. • Johns Hopkins Cystic Fibrosis Center. Science of cf: cftr function. Johns Hopkins Medicine. http://www.hopkinscf.org/what-is-cf-teen/science-of-cf-teen/cftr-teen/function-teen/. Published 2013. Accessed December 22, 2013. • US National Library of Medicine. Medline plus: glucose test. National Institute of Health. http://www.nlm.nih.gov/medlineplus/ency/article/003482.htm. Published June 6, 2012. Accessed January 6, 2014. • US National Library of Medicine. Medline plus: cystic fibrosis lung transplant. National Institute of Health. http://www.nlm.nih.gov/medlineplus/ency/article/000107.htm. Published August 8, 2010. Accessed January 6, 2014.
References • Chenoweth, R. H., Garwick, A. E., Lawler, M. R., et al. Normal and therapeutic nutrition 17th edition. NY, NY: Macmillan Publishing Company; 1986. • Feranchak, A. P., Quinton, H., Stallkings, V. A., et al. Evidence-based practice recommendations for nutrition-related management of children and adults with cystic fibrosis and pancreatic insufficiency. Journal of American Dietetic Association. May 2008; 108: 1-8. • Academy of Nutrition and Dietetics. Nutrition care manual: cystic fibrosis nutrition therapy. Eatright.org; 2014. Chicago, IL 2014. https://www.nutritioncaremanual.org/content.cfm?ncm_content_id=89545&highlight=cystic%20fibrosis. Accessed January 9, 2014. • Ittenbach, R. I., Olsen, I. E., Schall, J. I., et al. Evaluation of formulas for calculating total energy requirements of adolescents and children with cystic fibrosis. The American Journal of Clinical Nutrition. 2007; 85: 144-151. • Kains, D. and Wilchanski, M. Maintenance of nutritional status in patients with cystic fibrosis: new and emerging therapies. National Institute of Health. 2012; 6: 151-161. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3392141/. Accessed January 9, 2014. • Baker, R. D., Borowitz, D., and Stallings, V. Consensus report on nutrition for pediatric patients with cystic fibrosis. Journal of Pediatric Gastroenterology and Nutrition. 2010; 35: 246-259. http://www.cff.org/UploadedFiles/treatments/CFCareGuidelines/Nutrition/Consensus-Report-on-Nutrition-for-Pediatric-Patients-with-CF-JPGN-Sep-2002.pdf. Accessed January 11, 2014. • Pohl J. Nutrition in cystic fibrosis. Practical Gastroenterology Journal. 2010; 8:20-27.