Download

1 / 53
550 likes | 658 Vues
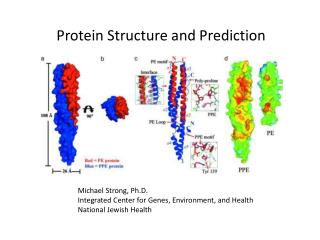
Forces and Prediction of Protein Structure. Ming-Jing Hwang ( 黃明經 ) Institute of Biomedical Sciences Academia Sinica. http://gln.ibms.sinica.edu.tw/. Sequence - Structure - Function.

E N D
Forces and Prediction of Protein Structure Ming-Jing Hwang (黃明經) Institute of Biomedical Sciences Academia Sinica http://gln.ibms.sinica.edu.tw/
Sequence - Structure - Function MADWVTGKVTKVQNWTDALFSLTVHAPVLPFTAGQFTKLGLEIDGERVQRAYSYVNSPDNPDLEFYLVTVPDGKLSPRLAALKPGDEVQVVSEAAGFFVLDEVPHCETLWMLATGTAIGPYLSILR
Sequence/Structure Gap Current (May 26, 2005) entries in protein sequence and structure database: • SWISS-PROT/TREMBL : 181,821/1,748,002 • PDB : 31,059 Sequence Structure
Structure Prediction Methods Homology modeling Fold recognition ab initio 0 10 20 30 40 50 60 70 80 90 100 % sequence identity
Levinthal’s paradox (1969) • If we assume three possible states for every flexible dihedral angle in the backbone of a 100-residue protein, the number of possible backbone configurations is 3200. Even an incredibly fast computational or physical sampling in 10-15 s would mean that a complete sampling would take 1080 s, which exceeds the age of the universe by more than 60 orders of magnitude. • Yet proteins fold in seconds or less! Berendsen
Energy landscapes of protein folding Borman, C&E News, 1998
Other factors • Formation of 2nd elements • Packing of 2nd elements • Topologies of fold • Metal/co-factor binding • Disulfide bond • …
Ab initio/new fold prediction • Physics-based (laws of physics) • Knowledge-based (rules of evolution)
1-microsecond MD simulation 980ns • villin headpiece • 36 a.a. • 3000 H2O • 12,000 atoms • 256 CPUs (CRAY) • ~4 months • single trajectory Duan & Kollman, 1998
Protein folding by MD PROTEIN FOLDING:A Glimpse of the Holy Grail? Herman J. C. Berendsen* "The Grail had many different manifestations throughout its long history, and many have claimed to possess it or its like". We might have seen a glimpse of it, but the brave knights must prepare for a long pursuit.
Massively distributed computing • SETI@home: • Folding@home • Distributed folding • Sengent’s drug design • FightAIDS@home • …
Massively distributed computing Letters to nature (2002) • engineered protein (BBA5) • zinc finger fold (w/o metal) • 23 a.a. • solvation model • thousands of trajectories each of 5-20 ns, totaling 700 ms • Folding@home • 30,000 internet volunteers • several months, or ~a million CPU days of simulation
Energy landscapes of protein folding Borman, C&E News, 1998
Protein-folding prediction technique CGU: Convex Global Underestimation - K. Dill’s group
Challenges of physics-based methods • Simulation time scale • Computing power • Sampling • Accuracy of energy functions
Structure Prediction Methods Homology modeling Fold recognition ab initio 0 10 20 30 40 50 60 70 80 90 100 % sequence identity
Flowchart of homology (comparative) modeling From Marti-Renom et al.
Fold recognition Find, from a library of folds, the 3D template that accommodates the target sequence best. Also known as “threading” or “inverse folding” Useful for twilight-zone sequences
Fold recognition (aligning sequence to structure) (David Shortle, 2000)
Reliability and uses of comparative models Marti-Renom et al. (2000)
Pitfalls of comparative modeling • Cannot correct alignment errors • More similar to template than to true structure • Cannot predict novel folds
Ab initio/new fold prediction • Physics-based (laws of physics) • Knowledge-based (rules of evolution)
From 1D 2D 3D Primary LGINCRGSSQCGLSGGNLMVRIRDQACGNQGQTWCPGERRAKVCGTGNSISAYVQSTNNCISGTEACRHLTNLVNHGCRVCGSDPLYAGNDVSRGQLTVNYVNSC seq. to str. mapping Secondary(fragment) Tertiary fragment assembly
One group dominates the ab initio (knowledge-based) prediction One lab dominated in CASP4
Some CASP4 successes Baker’s group