Download

1 / 36
360 likes | 368 Vues
Gene expression. Gene Expression. protein. RNA. DNA. Gene Expression. AAAAAAA. AAAAAAA. AAAAAAA. AAAAAAA. AAAAAAA. AAAAAAA. c. AAAAAAA. AAAAAAA. AAAAAAA. mRNA gene1. AAAAAAA. AAAAAAA. AAAAAAA. AAAAAAA. AAAAAAA. mRNA gene2. AAAAAAA. AAAAAAA. AAAAAAA. AAAAAAA. mRNA gene3.

E N D
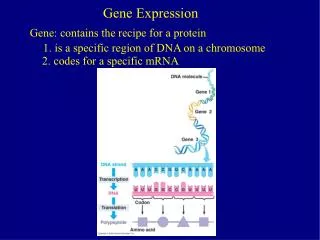
Gene Expression protein RNA DNA
Gene Expression AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA c AAAAAAA AAAAAAA AAAAAAA mRNA gene1 AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA mRNA gene2 AAAAAAA AAAAAAA AAAAAAA AAAAAAA mRNA gene3
Studying Gene Expression 1987-2013 cDNA Microarrays (first high throughput gene expression experiments) DNA chips (High density oligonucleotide microarrays ) RNA-seq (High throughput sequencing)
Microarray (DNAchip) – General idea A cDNA Synthesis+ Labeling Biological Sample RNA islolation cDNAs are hybridized to the array c c Millions of copies of probes (single strand oligonucleotides) are printed on each cell of the array B Image is scanned and analyzed Designing probes unique to each gene in the genome (more than one probe per gene) …………………….… …………………….. ………………………. DNA chip including thousand of different probes, complementary to different genes in the genome
Classical versus modern technologies to study gene expression • Classical Methods (Microarrays) • -Require prior knowledge on the RNA transcript • Good for studying the expression of known genes • High throughput RNA sequencing • Do not require prior knowledge • Good for discovering new genes (e.g. non coding RNAs) , • Good for studying splicing (alternative splicing events)
What can we learn from RNAseq? - Comparing the expression between two genes in the same sample • Comparing the expression between the same gene in different samples • Differential Expression
What can we learn from RNAseq? Comparing the expression between the same gene in different samples Example : Finding new markers for pluripotency (תאים ממוינים) (תאי גזע עובריים) Good markers for pluripotency -Show high differential expression Highly Expressed Lowly Expressed
How do to define differentially expressed genes? Comparing the expression between the same gene in different samples Sample X (Stem cell) Sample Y (Fibroblasts) Fold change (FC) = Ratio between the expression of the gene in sample X to the expression of the gene in sample Y Is fold change enough to evaluate the difference?
How do to define differentially expressed genes? If we only have one sample from each condition by chance we can get big differences that are completely not reliable !! A good experiment 3 Sample of Stem cell 3 Samples of Fibroblasts Y1 Y2 Y3 X1 X2 X3 NX1 NX2 NX3 NY1 NY2 NY3 (NX1 + NX2 + NX3 )/3 Fold change (FC) = (NY1 + NY2 + NY3 )/3
How do to define differentially expressed genes? 3 Sample of Stem cell 3 Samples of Fibroblasts FC Y1 Y2 Y3 X1 X2 X3 Gene A 100 80 90 35 25 30 3 Y1 Y2 Y3 X1 X2 X3 Gene B 180 70 20 65 15 10 3 Is fold change enough to evaluate the difference? NO
How do to define differentially expressed genes? To evaluate the significance of the difference we calculate the p-value In this case p-value = probability of the observed results assuming that there is no difference between the gene expression of the gene between the two samples (The p-value is calculated using a modified t-test) Significant p-value < 0.05 *** *** For real data we have to correct the p-value for multiple testing !! Why ? As we do many testing on many genes the probability to get a significant p-value by chance grows. There are different ways to correct for multiple testing : Bonferroni’s correction, False Discover Rate (FDR, q-value )
Volcano Plot Largest FC & Most Significant P-value To detect a differentially expressed genes we need to calculate the Fold Change (FC) and p-value * Possible candidates for being pluripotent markers Expression in stem cells versus fibroblasts * Here we calculate the –log (corrected p-value) high values denote highly significant results
NEXT… Clustering the data according to expression profiles Clustering organizes things that are close into groups. . Genes Expression in different human tissues Highly Expressed Lowly Expressed
Clustering the genes according to expression Gene Cluster A set of genes that have a similar expression pattern across tissues How do we define “similar expression pattern” ?
Clustering organizes things that are close into groupsWhat does it mean for two genes to be close? We need a mathematical definition of distance between the expression pattern of two genes 01 02 03 04 05 06 07 08 09 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Gene 1 Gene 2 Gene1= (E11, E12, …, E1N)’ Gene2= (E21, E22, …, E2N)’ We can used different approaches to define distance Approaches are roughly divided to “distance based” and “correlation based”
Calculating the distance between two expression patterns Gene1= (E11, E12, …, E1N)’ Gene2= (E21, E22, …, E2N)’ Euclidean distance (ED)= Sqrt of Sum of (E1i -E2i)2, i=1,…,N X1,Y1 Distance X2,Y2 When N is 100 we have to think abstractly Low Euclidean Distance High similarity
Calculating the correlation between two expression patterns r= 0.94 Pearson correlation coefficient Pearson’s correlation coefficient is a measure of the linear association between variables (ranges from -1 to 1) High correlation coefficient (1) High similarity
Distance and correlations can produce very different results Counts Euclidian distance= 1740 Pearson correlation= 0.9 High similarity Low similarity
WHY?What can we learn from the clustering genes? • Identify gene function • Set of genes with similar gene expression can infer similar function • Diagnostics and Therapy • A set of genes which differs in the gene expression can indicate a disease state
What can we learn from the clustering genes?-Identify gene function Similar expression between genes can suggest that: -The genes have similar function -The genes work together in the same pathway/complex
Example: Identifying genes that have similar function HnRNPA1 and SRp40 are not clear homologs based on blast e-value but have a very similar gene expression pattern in different tissues
Are hnRNP A1 and SRp40 functionally homologs ?? hnRNP A1 SF SF SF SF SF SF SF SF SF SF SF SF SRP40 Yes both genes are splicing factors !
Example: Identify genes that work together in the same pathway/complex Counts Transcription Factor TF Long non-coding RNA
What can we learn from the clustering genes?-Diagnostic and Therapy How can gene expression help in diagnostics?
A molecular signature of metastasis in primary solid tumors Samples were taken from patients with adenocarcinoma. hundreds of genes that differentiate between cancer tissues in different stages of the tumor were found. The arrow shows an example of a tumor sample that was not detected correctly by histological or other clinical parameters. Ramaswamy et al, 2003 Nat Genet 33:49-54
How can gene-expression help in diagnostics ? Genes RESEARCH QUESTION Defining the subclass of lymphoma for providing most suitable treatment 42 diffuse large B-cell lymphoma (DLBCL), 9 follicular lymphoma (FL) and 11 chronic lymphocytic leukemia (CLL). Cancer type HERE we want to cluster the patients so we can characterize a sub class of cancer based on the gene expression profile
HOW?Different clustering approaches • Unsupervised (למידה בלתי מונחית) - Hierarchical Clustering - K-means - Principal component analysis (PCA) • Supervised Methods(למידה מונחית) -Support Vector Machine (SVM)
Unsupervised approachesfor diagnostic based on expression data We define the number of classes Number of classes non provided
Unsupervised approachesfor diagnostic based on expression data Each dot represents one sample and is colored according to the group the sample belongs to Important the colors are assigned only after the analysis and are not part of the algorithm ! They help visualizing the data.
How to interpret a PCA? PCA is a way to simplify a large and complex data set into a smaller, more easily understandable data set. In PCA we start in reducing the large number of features (in our case the gene expression) to a smaller number of variable that are most informative. These features are called “components” and describe a linear combination of the original features. In this plot we see the scattering of the samples based on the two most informative components (1st and 2nd components) that capture most of the variance in the data.
Supervised approachesfor diagnostic based on expression data Support Vector Machine SVM
How can gene-expression help in diagnostics ? Patients BRCA2 BRCA1 DATA Microarray expression of all genes from two types of breast cancer patients (BRCA1 and BRCA2) Red =high expression Green=low expression Genes Clustering is not trivial!!!
SVM would begin with a set of samples from patients which have been diagnosed as either BRCA1 (red dots) or BRCA2 (blue dots). Each dot represents a vector of the expression pattern taken from the microarray experiment of a patient. NOTE!! The dots are shown in a 2-dimension plot for demonstration only.
? How do SVM’s work with expression data? The SVM is trained on data which was classified based on histology. After training the SVM to separated the BRCA1 from BRAC2 tumors given the expression data, we can then apply it to diagnose an unknown tumor for which we have the equivalent expression data .