Download
1 / 69
690 likes | 857 Vues
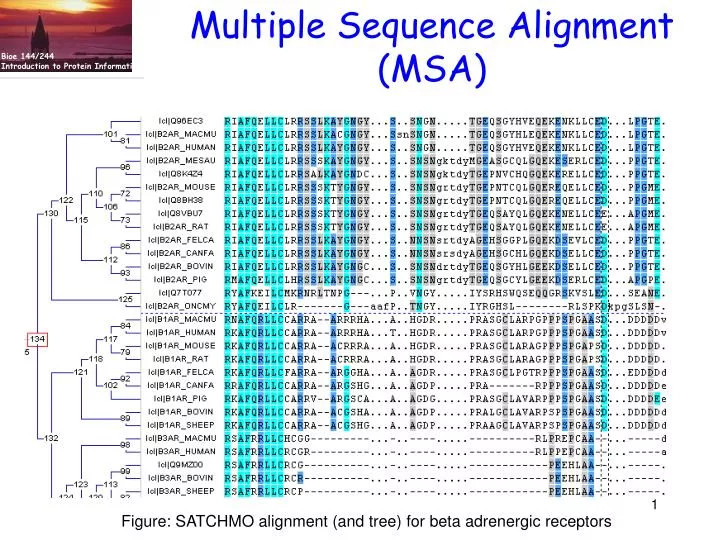
Multiple Sequence Alignment (MSA). Figure: SATCHMO alignment (and tree) for beta adrenergic receptors. Source material. Creighton, Proteins . Chapter 3: Evolutionary and Genetic Origins of Protein Sequences. pp 105-137

E N D
Multiple Sequence Alignment (MSA) Figure: SATCHMO alignment (and tree) for beta adrenergic receptors
Source material • Creighton, Proteins. Chapter 3: Evolutionary and Genetic Origins of Protein Sequences. pp 105-137 • Presents the evolutionary mechanisms by which protein families develop new folds and functions, and the functional and structural roles of individual positions in molecules. Foundational material for all aspects of protein informatics. • Recommended: • Thompson, Plewniak and Poch, “A comprehensive comparison of multiple sequence alignment programs” NAR 1999 • Presents the BAliBASE benchmark alignment dataset and results of different methods. Since it was published almost 10 years ago, it doesn’t include methods developed more recently, but it’s still relevant. BAliBASE was the first benchmark for multiple alignment accuracy and is still in use. • Geoffrey Barton, in Bioinformatics: A practical guide to the analysis of genes and proteins (course text). Chapter 12: Creation and Analysis of Protein Multiple Sequence Alignments. pp 325-336 • Excellent overview of many issues in multiple sequence alignment and interpretation by one of the top people in the field.
What is an MSA? • An MSA is an assertionof homology across >2 nucleotide or amino acid sequences. An MSA asserts that: • Sequences have a common ancestor • Characters in columns have descended from an ancestral character • The exception: indel characters represent deletion/insertion • The impact of these assertions not being supported by the alignment may be large or small, depending on the intended use • alignment editing/masking is employed to ameliorate conflicts between the data and assumptions • An MSA is a matrix M, with c columns and r rows. • Mi,j = the character for sequence i at column j. • In an MSA for proteins, characters are drawn from the set {a, c, d, e, …y, A, C, …Y, ., -} • Lower-case characters may have different meanings from upper-case characters • Dot (.) and dash (-) are indel characters (and may have different meanings from each other in some cases
Uses of sequence alignment Phylogenetic tree Active and binding site prediction GFKLP GYKLP GFRVP GF-LP Homology Models And more… Profiles/HMM construction Domain Prediction Substitution Matrices Function prediction by phylogenomic analysis Secondary structure prediction Subfamily identification …
Domain shuffling and gene fusion/fission events Leucine-Rich Repeat (LRR) Toll-Interleukin Receptor (TIR) domain Sequences containing domains found in different types of proteins complicate homolog detection and function prediction
Protein superfamilies exhibit structural and functional diversity
Local, glocal and global-global • Local-local • Global-local (aka glocal) • Global-global Best for boosting remote homolog detection, identification of evolutionary domains. Default protocol of BLAST and PSI-BLAST. Global to the query, potentially local to the hit. Best for gathering homologs to a structural domain. Restrict sequences to those appearing to have the same domain architecture. Recommended for phylogenomic inference of molecular function. Default protocol of FlowerPower and PhyloBuilder.
SAM a2m format • The UCSC SAM HMM software uses a specialized format for alignments, to describe how a sequence was emitted by an HMM (or, equivalently, aligns to an HMM). a2m format MSA columns are of two types: • Columns consisting of upper-case characters and dashes correspond to nodes in the HMM representing the consensus structure • Dashes are placed to indicate passage through an HMM skip/delete state • I.e., a dash indicates a sequence does not have the consensus structure at that position • Columns consisting of lower-case characters and dots correspond to residues emitted in HMM insert states, representing inserts between positions in the consensus structure • Dots are inserted post-hoc so that all sequences in the MSA have the same number of characters. • Dots in one sequence indicate that another sequence has inserted characters using an insert state at that position
Some alignments are easy Note: no gaps, high sequence identity.
Caveats • Sequence “signal” guides the alignment • If the signal is weak, the alignment is likely to be poor • As proteins diverge from a common ancestor, their structures and functions can change • All methods perform poorly when pairs are included with <25% identity • Even structural superposition can be challenging! • Unequal numbers of repeats, domain shuffling, large insertions or deletions introduce significant alignment errors • Take care in selecting sequences • See Geoff Barton’s recommendations on iterative alignment editing and re-alignment (from text)
Tree and alignment accuracy • Alignment methods are assessed for accuracy relative to structural alignment • Tree methods are assessed via simulation studies (which do not adequately assessed the kind and degree of variability observed in protein families)
Structural alignment is the gold standard • Structural superposition of two PDB structures provides correspondences/equivalences between residues • Since primary sequence diverges more rapidly than 3D structure, structural alignment is the gold standard against which sequence alignment is assessed • Not all structural aligners agree on all pairs • However, clearly superposable pairs (within 2.5 Angstroms) are normally agreed upon by structural aligners • Example structural aligners include: CE, DALI, VAST, Structal
Structural alignment example ID EC Function 1E9Y 3.5.1.5 Urease 1J79 3.5.2.3 Dihydroorotase Identity 9.8% Equivalent Residues 40%
VAST Structural Alignment at NCBI Type in the PDB structure ID of interest. http://www.ncbi.nlm.nih.gov/Structure/VAST/vast.shtml
Protein superfamilies exhibit structural and functional diversity
Then select PDB structures for which you want to see a structural alignment
VAST alignment of selected structures VAST multiple alignment based on structure superposition: Non-equivalent positions are in lower-case 1SN4 Scorpion neurotoxin (colored manually using PhyloFacts JMOL viewer to display non-equivalent positions)
Sequence and structural divergence are correlated Accuracy of sequence alignment relative to structural alignment Left three columns show results of structural alignment %ID: Structure pairs have been placed into bins based on sequence identity given the structural alignment #pair: number of pairs in each bin %Superpos: percent positions that are within ~3Angstroms RMSD (between backbone C-alpha carbons) Right three columns give Cline Shift scores for pairwise sequence alignments relative to the structural alignment. The best CS score possible is 1; negative scores indicate incorrect over-alignment with very few (or no) correctly aligned residue pairs.
SATCHMO: Simultaneous Alignment and Tree Construction using Hidden Markov mOdels Xia Jiang Nandini Krishnamurthy Duncan Brown Michael Tung Jake Gunn-Glanville Bob Edgar Edgar, R., and Sjölander, K., "SATCHMO: Sequence Alignment and Tree Construction using Hidden Markov models," Bioinformatics. 2003 Jul 22;19(11):1404-11
SATCHMO motivation • Structural divergence within a superfamily means that… • Multiple sequence alignment (MSA) is hard • Alignable positions varies according to degree of divergence • Current MSA methods not designed to handle this variability • Assume globally alignable, all columns (e.g. ClustalW)… • Over-aligns, i.e. aligns regions that are not superposable • …or identify and align only highly conserved positions (e.g., SAM software with HMM “surgery”) • Challenge • Different degrees of alignability in different sequence pairs, different regions • Masking protocols are lossy: loop regions may be variable across the family but may be critical for function!
SATCHMO algorithm • Input: unaligned sequences • Initialize: a profile HMM is constructed for each sequence. • While (#clusters > 1) { • Use profile-profile scoring to select clusters to join • Align clusters to each other, keeping columns fixed • Analyze joint MSA to predict which positions appear to be structurally similar; these are retained, the remainder are masked. • Construct a profile HMM for the new masked MSA } • Output: Tree and MSA
Assessing sequence alignment with respect to structural alignment Xia Jiang Duncan Brown Nandini Krishnamurthy
Multiple alignment methods • Main classes of methods • Progressive: • Once a sequence is aligned, that alignment will not change • Sequences are typically (but not always) aligned using some pre-specified order (e.g., based on a guide tree); • Examples: ClustalW, SATCHMO • Note: not all progressive alignment methods use a guide tree to determine the order in which sequences are aligned • Iterative: • Alignments are refined during each iteration; allows sequences to adjust their alignments • Examples: FlowerPower (if sequences are realigned in each iterations), • Mixed (including progressive and iterative approaches) • Example: MUSCLE, ProbCons • HMM approaches (SAM buildmodel, and align2model)
Alignment editing • Alignment editing • Based on the intended use of the alignment • Removing outlier sequences or fragments • Removal of non-homologs important for profile/HMM construction • Some tree methods are sensitive to how gaps are handled • Including fragmentary matches can have unexpected impact • Removing columns • Masking variable regions is important for phylogenetic tree construction • Deleting very gappy regions can be critical for HMM performance • Making the alignment non-redundant • May be important for some profile/HMM methods • Sequence weighting can sometimes accomplish the same objective • E.g., UCSC SAM w0.5 handles these issues for the user
Master-slave alignment • One sequence is the “master” • Other sequences (“slaves”) are aligned to the master • Examples: BLAST, PSI-BLAST • You can also get this result using HMM methods • Construct an HMM (e.g., using w0.5) • Align putative homologs to the HMM (e.g., using align2model)
Phylogenomic inference of protein function FlowerPower for clustering sequences sharing same domain architecture (or manual selection) Re-align using preferred method FlowerPower provides MUSCLE realignment Mask variable or gappy columns Construct tree Or, use the PhyloBuilder software Task 1: Phylogenomic inference http://phylogenomics.berkeley.edu/phylobuild/
To predict structural domains Include locally aligning proteins in MSA Best: construct HMM for global homologs; align local matches using local-local alignment (SAM -sw 2) Examine alignments displayed in BLAST and PSI-BLAST Submit sequences to domain prediction servers Task 2: MSA construction/analysis for remote homolog detection and structure prediction DAPHNE analysis of K+ channel alignment (including partial homologs)
Compare results from BLAST and domain prediction servers to refine domain boundaries PFAM: TIR ends at 142 (also finds LRR; see next slide) SMART: TIR ends at 145 PhyloFacts: TIR ends at 156 (finds LRR, but not NB-ARC) BLAST: N-terminal match ends at 169 PhyloFacts results NCBI CDD SMART results BLAST hit NCBI CDD includes models from SMART and PFAM. Different resources represent domains differently (often crop domains to highly conserved regions).
Using multiple alignment as a tool for structure prediction Since structure is (largely) conserved, sequence homologs from the MSA can also be submitted to structure prediction servers Alignment analysis to identify (apparently) critical residues for the family can be used to judge possible homologs in PDB (i.e., check the alignment of the PDB sequence against the consensus for the family) Predicting Protein Structure Using Hidden Markov Models. Proteins: Structure, Function and Genetics. Suppl. 1. (1997), Karplus, Sjölander, Barrett, Cline, Haussler, Hughey, Holm and Sander.
Using sequences from an MSA for structure prediction Homolog identification Since structure is (largely) conserved, sequence homologs from the MSA can also be submitted to structure prediction servers Some of these homologs may function as intermediate sequences between the target and a solved structure Servers to try: 3d-pssm, PHYRE, Superfamily, PhyloFacts Cautions/comments: Restrict sequence homologs to the regions where they align to the target! Not all servers are kept current with PDB, so not all structures will be represented by a server
For an individual domain (found in multi-domain proteins) Cluster and align sequences using master-slave approach (PSI-BLAST, SAM-T2K or FlowerPower) Re-align subsequences using preferred alignment method Edit to remove outlier sequences Phylogenetic tree construction Mask variable or gappy columns Profile/HMM construction Mask very gappy columns Task 3: Domain-specific alignment
To reconstruct a species tree Select orthologs from many species (manual) Align using preferred alignment method Alignment masking N.B. Don’t rely on results from just one protein or gene family! Task 4: Species tree construction
Alignment Editing Belvu allows: • Coloring columns according to characteristics • Changing sequence order (by %ID, tree topology) • Deleting columns (specified range or characteristics) • Deleting sequences individually, or according to characteristics (fraction gaps, low %ID) • And more…
BAliBASE Julie Thompson, Frédéric Plewniak and Olivier Poch (1999) Bioinformatics, 15, 87-88
Motivation (paraphrased from the authors) • The alignment of protein sequences is a crucial tool in molecular biology and genome analysis. • Historically, the quality of new alignment programs has been compared to previous methods using a small number of test cases selected by the program author. [italics mine] • Comparisons have been done using a set of alignments selected from structural databases. • These databases assemble proteins into homologous families, but alignments are not classified specifically for the systematic evaluation of multiple alignment programs. http://bips.u-strasbg.fr/en/Products/Databases/BAliBASE/
BAliBASE Motivation (cont’d) • It has been shown (McClure et al., 1994) that the performance of alignment programs depends on • the number of sequences, • the degree of similarity between sequences and • the number of insertions in the alignment. • Other factors may also affect alignment quality • length of the sequences • existence of large insertions • N/C-terminal extensions • over-representation of some members of the protein family. • We have constructed BAliBASE (Benchmark Alignment dataBASE) containing high-quality, documented alignments to identify the strong and weak points of the numerous alignment programs now available. http://bips.u-strasbg.fr/en/Products/Databases/BAliBASE/
Reference alignment construction • The sequences included in the database are selected from alignments in either the FSSP or HOMSTRAD structural databases, or from manually constructed structural alignments taken from the literature. • When sufficient structures are not available, additional sequences are included from the HSSP database (Schneider et al., 1997). • The VAST Web server (Madej, 1995) is used to confirm that the sequences in each alignment are structural neighbours and can be structurally superimposed. • Functional sites are identified using the PDBsum database (Laskowski et al., 1997) • Alignments are manually verified and adjusted, in order to ensure that conserved residues are aligned as well as the secondary structure elements. http://bips.u-strasbg.fr/en/Products/Databases/BAliBASE/
Determining “core blocks” • A frequent problem encountered when using reference alignments has been the effect of ambiguous regions in the sequences which cannot be structurally superposed. • Very distantly related sequences often have only short conserved motifs in long regions of low overall similarity. These regions can only be aligned arbitrarily in the reference and may lead to a bias in the comparison of programs. • In BAliBASE, we have annotated the core blocks in the alignments that only include the regions that can be reliably aligned. • The blocks exclude regions where there is a possibility of ambiguity in the alignment. This may be an important factor affecting the significance of statistical comparisons of alignment programs. http://bips.u-strasbg.fr/en/Products/Databases/BAliBASE/
Reference alignments • Reference 1 contains alignments of < 6 equidistant sequences (i.e. the pairwise identity is within a specified range). All the sequences are of similar length, with no large insertions or extensions. • Reference 2 aligns up to three "orphan" sequences (less than 25% identical) from reference 1 with a family of at least 15 closely related sequences. • Reference 3 consists of up to 4 subgroups, with less than 25% residue identity between sequences from different groups. The alignments are constructed by adding homologous family members to the more distantly related sequences in reference 1. • Reference 4 is divided into two sub-categories containing alignments of up to 20 sequences including N/C-terminal extensions (up to 400 residues), and insertions (up to 100 residues). http://bips.u-strasbg.fr/en/Products/Databases/BAliBASE/