Download

1 / 22
220 likes | 312 Vues
Phylogentic Tree Construction. (Lecture for CS397-CXZ Algorithms in Bioinformatics) April. 2, 2004 ChengXiang Zhai Department of Computer Science University of Illinois, Urbana-Champaign. Introduction.

E N D
Phylogentic Tree Construction (Lecture for CS397-CXZ Algorithms in Bioinformatics) April. 2, 2004 ChengXiang Zhai Department of Computer Science University of Illinois, Urbana-Champaign
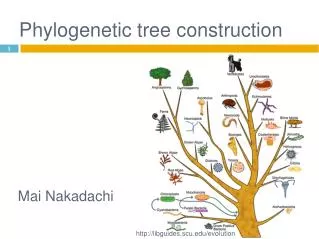
Introduction • Phylogenetic tree: A tree with sequences as leaves that reflect evolutionary relationship • Formal properties • Binary • Rooted or unrooted • Edge length reflects the amount of evolutionary divergence. • Contruction methods (all related to clustering) • Similarity/distance based (bottom up construction) • Maximum parsimony (search for the right tree) • Probabilistic models (modeling a tree)
Similarity-based Methods • Unweighted Pair Group Method using Arithmetic Averages (UPGMA) • Essentially average-link clustering • Node height (Ck) = ½ dij, dij is the distance of the two children of Ck • Desirable properties of tree • Molecular clocks (edge lengths): Equal edge length to the leaves from the same node (tree shows the time) • Additivity: Edge lengths are additive if the distance between any pair of leaves is the sum of the lengths of the edges on the path connecting them. (tree shows “changes”) • UPGMA can guarantee “molecular” but not necessary “additivity”.
Neighbor-Joining • Adjust the distances • Dij = dij –(ri +rj), ri is the average distance of i to all other nodes • Guarantees minimum Dij=> neighbors • Alternative cluster distance function • Suppose i and j are a pair of neighbors, replacing them with a new node k • Define dkm = ½ (dim + djm –dij) for any other node m • This guarantees additivity • Finally, the edge length is dik = ½ (dij +rj -rj), djk =dij –dik, for joining k to i and j. • Used in ClustalW
Neighbor-Joining: Example A Original distance matrix 3 r 13.5 15.5 13.5 18.5 1 5 2 3 B C 6 Adjusted distance matrix 8-(13.5+15.5) D Original (true) tree
Neighbor-Joining: Example (cont.) (8-(15.5-13.5))/2=3 Intermediate distance matrix C r 13 15 20 A 3 4 F 11 9 B 5 D 4-(13+15) (8+(15.5-13.5))/2=5 Adjusted distance matrix Original distance matrix dFC=(dAC+dBC-dAB)/2=4 A 3 C 3 F 1 8 B 5 root 6 D
Maximum Parsimony maximum parsimony principle: the principle that the most accurate phylogenetic tree is one that is based on the fewest changes in the genetic code.
0 0 0 1 2 3 4 5 6 7 8 9 10 1 - A G G G T A A C T G 2 - A C G A T T A T T A 3 - A T A A T T G T C T 4 - A A T G T T G T C G
0 3 0 3 0 3 1 2 3 4 5 6 7 8 9 10 1 - A G G G T A A C T G 2 - A C G A T T A T T A 3 - A T A A T T G T C T 4 - A A T G T T G T C G
G T C A C G C T A C G T A C C 3 4 1 - G 2 - C 3 - T 4 - A 3 3
0 3 2 0 3 2 0 3 2 1 2 3 4 5 6 7 8 9 10 1 - A G G G T A A C T G 2 - A C G A T T A T T A 3 - A T A A T T G T C T 4 - A A T G T T G T C G Informative Site=discriminative site
0 3 2 2 0 3 2 2 0 3 2 1 1 2 3 4 5 6 7 8 9 10 1 - A G G G T A A C T G 2 - A C G A T T A T T A 3 - A T A A T T G T C T 4 - A A T G T T G T C G
G A 2 A G A G A 2 A G A A G 1 A G A 4 1 - G 2 - A 3 - A 4 - G
1 2 3 4 5 6 7 8 9 10 1 - A G G G T A A C T G 2 - A C G A T T A T T A 3 - A T A A T T G T C T 4 - A A T G T T G T C G 0 3 2 2 0 3 2 2 0 3 2 1
1 2 3 4 5 6 7 8 9 10 1 - A G G G T A A C T G 2 - A C G A T T A T T A 3 - A T A A T T G T C T 4 - A A T G T T G T C G 0 3 2 2 0 1 1 1 1 3 14 0 3 2 2 0 1 2 1 2 3 16 0 3 2 1 0 1 2 1 2 3 15
Probabilistic Approaches • Basic idea: • Tree= Generative probabilistic model, e.g., an n-leaf tree defines a model p(X1, …,Xn) • Data: sequences {s1, …, sn} • Choose the tree according to • Maximum Likelihood: p(Data|Tree) • Maximum A Posterior (Bayesian): p(Tree|Data) • Model evolution more directly • Computationally expensive
Detailed View of Probabilistic Models The tree on the left defines the following probabilistic model: x5 t4 x4 t2 t3 Basic evolution model: p(x|y,t)=prob of x arising from an ancestral sequence y over an edge of length t t1 x2 x1 Decompose the sequence: “Independence Assumption”: x3 Decompose the time: “Markovian Assumption” • “Primitive Evolution Model”: p(a|b,t) • - Nucleotides: Jukes-Cantor model • - Amino acids: PAM
The Jukes-Cantor model A C G T R= S(t)= Solutions: rt = (1+3e4t)/4, st = (1 e4t)/4.
Computing the Likelihood With Parents Known: x5 t4 x4 t2 t3 But We don’t know the parents… t1 x2 x1 x3
Handling the Hidden Nodes • We must sum up over all the hidden ancestral nodes • Felsenstein’s algorithm for likelihood: Compute the sum in a bottom up fashion • Start from leaves • Compute the parent node based on children nodes
Maximizing the Likelihood • Easy for small number of sequences • Generally complex for large number of sequences • Many solutions: • EM • Gradient descent • Sampling • Metropolis sampling • Accept a new tree if P(new-tree)>= P(old-tree) • Accept a new tree with prob. P(new-tree)/P(old-tree) if p(new-tree)<p(old-tree)
More realistic evolutionary models • Allowing different rates at different sites • Using a prior (e.g., gamma) to regular the different rates • Hidden Markov models • Evolutionary models with gaps • Tree HMMs