Download

1 / 49
520 likes | 884 Vues
SECRETARIA DE ESTADO DE SAUDE DO DISTRITO FEDERAL HOSPITAL REGIONAL DA ASA SUL UNIDADE DE PEDIATRIA. FISIOLOGIA DA HEMOSTASIA. www.paulomargotto.com.br Brasília, 18/4/2010. Melina Swain. INTRODUÇÃO. Hemostasia

E N D
SECRETARIA DE ESTADO DE SAUDE DO DISTRITO FEDERAL HOSPITAL REGIONAL DA ASA SUL UNIDADE DE PEDIATRIA FISIOLOGIA DA HEMOSTASIA www.paulomargotto.com.br Brasília, 18/4/2010 Melina Swain
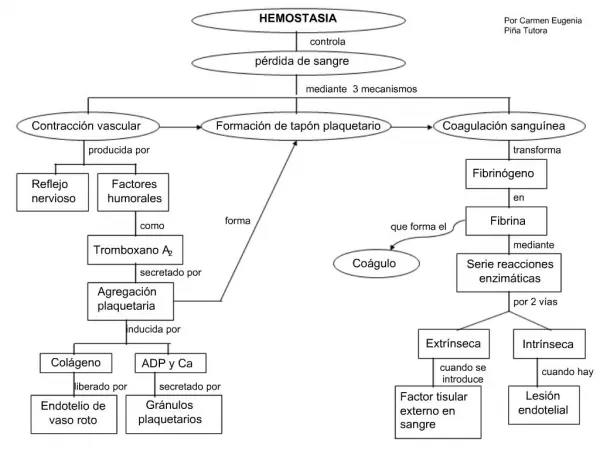
INTRODUÇÃO • Hemostasia • Mecanismo que garante o equilíbrio entre a coagulação excessiva (trombose) e hemorragia. • Dependente das atividades realizadas • Parede vascular (endotélio) • Plaquetas • Sistema de coagulação • Fibrinólise
PLAQUETAS • Pequenos fragmentos citoplasmáticos anucleados • Formados a partir da fragmentação citoplasmática dos megacariócitos • Forma discóide • circulam por 7 a 10 dias, sem interagir com outras plaquetas ou outras células do sangue • Quando expostas a agente agonista → passam de estado não adesivo para condição adesiva • Na hemostasia as plaquetas desempenham as funções de adesão, secreção, agregação, e atividade pró- coagulante
Adesão e ativação • Ocorrem nos locais de lesão vascular • Processo de várias etapas • Envolvem • Interação plaquetária com a matriz extracelular subendotelial, onde se encontram o colágeno e fator de Von Willebrand • Tecido subendotelial exposto • Fator de Von Willebrand sofre alteração da sua configuração →capaz de interagir com receptor plaquetário → glicoproteína (GP) Ib/V/IX • Essa ligação faz as plaquetas passarem mais lentamente sobre a superfície lesada→ interagindo com receptor para colágeno, a GP VI • GP VI - induz processos intracelulares de sinalização, provocando ativação de integrinasplaquetárias como: • αIIbβ3 (GP IIB/IIIa) ou α2β1 ( GPIa/IIa) • Se ligarão à matriz extracelular→ligação firme das plaquetas à parede vascular lesada → monocamada plaquetária
Trombo branco ou tampão plaquetário • Plaquetas ativadas →reações • Mudança de forma • Secreção granular • Agregação mediada por integrinaαIIb/β3 Recrutamento de plaquetas adicionais Agentes estimulantes mediadores ADP/ATP; tromboxano (TxA2) Mediado por trombina- produzida na superfície das plaquetas ativadas Produzidos ou liberados quando se inicia a adesão plaquetária e quando já ocorreu certo grau de ativação Através de mecanismo “flip-flop”- fosfatidilserina expressa na membrana celular – superfície fofolipídica com carga elétrica negativa Possibilita desenvolvimento local da “cascata” da coagulação
Secretam seu conteúdo quando há aumento da concentração intracitoplasmática de cálcio nas plaquetas ativadas • Três tipos de grânulos • Alfa • Denso • Lisossomas Grânulos alfa Contém proteínas como o fator de crescimento derivado de plaquetas (PDGF) , fibrinogênio e fator de Von Willebrand. P-seletina e GP IIb/IIIa (proteínas de membrana): translocadas para a membrana citoplasmática quando o grânulo sofre processo de exocitose→ ligação dos fatores de coagulação, que após interações culmina na geração local de trombina Grânulos densos Armazenam ADP e 5-hidroxitriptamina ou serotonina ( 5-HT). Como as plaquetas apresentam receptores específicos para ADP e 5HT, a secreção destas substâncias aumenta a ativação plaquetária (retroalimentação positiva) Lisossomas Estocam enzimas hidrolíticas, que contribuem para remodelação vascular.
Plaquetas ativadas • Tromboxano A2 (Tx A2 ) produzido a partir do ácido araquidônico da membrana celular, após sua secreção atua como agonista plaquetário → papel na ativação sinérgica das plaquetas no local de formação do trombo. • GPIIb/IIIa – medidor do processo de agregação plaquetária - na membrana plasmática e nos grânulos alfa das plaquetas não ativadas • GPIIb/IIIa não expressa atividade de ligação, quando as plaquetas estão em estado não estimulado. Com ativação da plaqueta sofre alteração conformacional, passando a ter capacidade de ligar-se ao fibrinogênio e fator de Von Willebrand • Vários sítios de ligação com a GPIIb/IIIa ativada e induzem agregação plaquetária ao promover interligação de plaquetas adjacentes • Ativação da GPIIb/IIIa é o resultado final de varias vias de sinalização, também chamada de “via final comum” • GPIIb/IIIa pode se ligar ao fibrinogênio e fator de Von Willebrand na dependencia do estresse de cisalhamento local: • estresse elevado → fator de Von Willebrand • Estresse reduzido→fibrinogênio
MECANISMO DE COAGULAÇÃO • Processo de coagulação • Série de reações enzimáticas que ocorrem sobre uma superfície fosfolípidica→ a membrana citoplasmática de uma célula ativada • Ativação do mecanismo de coagulação tem inicio por duas vias diferentes • Via intrinseca • via extrinseca • Evento central é geração de trombina • Quatro fatores • II • VII • IX • X • Produzidos no fígado Carboxil depende de vit.K, que cataliza reação de adição do grupo carboxil em residuo de ácido glutâmico→ dois resíduos de ácido caboxiglutâmico. Duplo resíduo de carboxiglutâmico +cálcio promove ligação do fator à superficiefosfolipidicapossibita a atvidade biológica do fator ativado Dependem de vitamina K para expressar suas funções biológicas
Via intrinseca • Fator XII se liga a fibrila de colágeno sub endotelial, que fica exposta após lesão do vaso sanguíneo • Ao unir-se à fibrila de colágeno→formação de complexo com o cininogênio de alto peso molecular (HMWK) e com a pré-calicreína (PK)→ ativando o fator XII→FXIIa • FXIIa ativa o fator XI, que por sua vez ativa outros fatores de coagulação • FXIa também ativa a pré-calicreína e a transforma em calicreína→ acelera a ativação do FXII
Via extrínseca • Depende de fator não circulante • Fator tecidual (FT)→ lipoproteína que faz parte das membranas celulares. • Lesão endotelial →exposição do FT • Ativação do FVII na presença de cálcio • Formação de complexo com a participação do FT, FVII e cálcio
Via comum Ponto de convergência das duas vias • FVIIa → via extrínseca • FXIa → via intrínseca • ativação dos fatores IX e X com a participação do fator VIII • Formação de complexo onde os FIX e FX estão ligados pelo cálcio a fofolipídeos de membrana celular • FXa - consequência das duas vias que iniciam a coagulação • Geração da trombina→ agente principal da coagulação
Geração da trombina • Ação do FXa sobre a protrombina (FII) • Participação do FV e cálcio na presença do fosfolipídio da membrana de qualquer célula • Quando acontece na membrana plaquetária a geração da trombina é acelerada milhares de vezes • Trombina: ação em múltiplos pontos no processo hemostático • Ativação das plaquetas no trombo hemostático • Ativa os fatores V, VIII, XIII • Atua sobre fibrinogênio • Liberação de fibrinopeptídios A e B → gerando monômeros de fibrina, que ligam-se longitudinalmente e lateralmente→ formando os polímeros de fibrina • Fibrina é solúvel pois monômeros são ligados por pontes de hidrogênio • Atuação do FXIIIa que confere insolubilidade à fibrina
ANTICOAGULAÇÃO • MECANISMOS REGULADORES DA COAGULAÇÃO • Reações bioquímicas da coagulação do sangue • Reguladas para evitar ativação excessiva do sistema, formação inadequada de fibrina, e, oclusão vascular • Atividade das proteases atuantes na ativação da coagulação é regulada por numerosas proteínas inibidoras→ anticoagulantes naturais • Principais inibidores fisiológicos da coagulação • TFPI→ “tissue factor pathway inhibitor” • PC→ proteína C • PS→ proteína S • AT→ antitrombina
Complexo FVIIa/FT • Atua sobre dois substratos principais – Ativação de FIX e FX Reações reguladas pelo inibidor da via do fator tecidual (TFPI) • TFPI • Proteína produzida pelas células endoteliais • Apresentam 3 domínios do tipo “Kunitz” • 1º domínio→ liga-se e inibe o complexo FVIIa/FT • 2º domínio→ liga-se e inibe o FXa • Ativação direta do FX é regulada negativamente de modo rápido na presença do TFPI→ limita produção do FXa e FIXa • Ligação do FXa - necessária para inibição do complexo FVIIa/FT pelo TFPI.
Proteína C ativada (PCa) • Ativada pela ligação ao seu receptor no endotélioEPCR (“endothelial PC recptor”) após ligação da trombina ao receptor endotelial trombomodulina (TM) • PCa inibe a coagulação clivando e inativando os FVa e FVIIa • Processo potencializado pela PS • Atua como cofator não enzimático nas reações de inativação • Função procoagulante quando gerada em excesso • Potente anticoagulante, em pequena quantidade → sua ligação à TM endotelial é o ponto chave para ativação da via inibitória da PC
AT (anteriormente denominada AT III) • Inibidor primário da trombina • Efeito inibitório sobre diversas enzimas da coagulação • FIXa, FXa, FXIa • Acelera a dissociação do complexo FVIIa/FT e impede sua reassociação • Elimina qualquer atividade enzimática procoagulante excessiva ou indesejável • Molécula de heparan-sulfato • Proteoglicana presente na membrana das células endoteliais • Acelerador das reações catalisadas pela AT • Atividade inibitória da AT é potentemente acelerada pela heparina • Polissacarídeo linear estruturalmente similar ao heparan-sulfato
SISTEMA PLASMINOGÊNIO/PLASMINA (Sistema fibrinolítico) Fibrinólise • Degradação da fibrina mediada pela plasmina • Composto por diversas proteínas (proteases séricas e inibidores) • Regulam a geração de plasmina→ enzima ativa produzida a partir de pro-enzima inativa (plasminogênio) • Função: degradar a fibrina e ativar metaloproteinases de matriz extracelular • Outras funções do sistema plasminogênio/plasmina em outros processos • Remodelagem da matriz extracelular • Crescimento e disseminação tumoral • Cicatrização • Infecção
Dois ativadores fisiológico do plasminogênio • Ativador do plasminogênio do tipo tecidual (t-PA) • Ativador do plasminogênio tipo uroquinase (u-PA) Formação de seríno-protease ativa → Plasmina • Plasmina não degrada apenas a fibrina, também o fibrinogênio, FV e FVIII • Em condições fisiológicas fibrinólise → processo altamente específico para fibrina • Ativação localizada e restrita, não sistêmica • Função de remover o excesso de fibrina do intravascular de modo equilibrado
TROMBOFILIAS HEREDITÁRIAS
Trombofilia Definição • Tendência à trombose decorrente de alterações hereditárias ou adquiridas da coagulação ou da fibrinólise, que levam a um estado pró-trombótico
Classificados em dois grupos distintos : • I - indivíduos que desenvolveram fenômenos tromboembólicos associados a doenças neoplásicas, traumas, imobilização prolongada no leito ou devido à presença de anticorpo antifosfolipídeo. • II - indivíduos com tendência trombótica geneticamente determinada, independente dos fatores de risco exógenos, sendo definido como trombofilia hereditária ( TH ).
Mecanismo de hipercoguabilidade • Geração de trombina - evento básico na hemostasia sanguínea. • Papel central nos dois sistemas de hemostasia • Efeito anticoagulante observado com baixos níveis plasmáticos; • Tendência para coagulação paulatinamente observada com o aumento da concentração plasmática.
Causas de TH Frequentes ou bem estabelecidas • deficiência de antitrombina III (AT) • deficiência de proteína C (PC) • deficiência de proteína S (PS) • resistência à PCa (FVQ 506 / Fator V Leiden) • hiperhomocisteinemia
Raras ou não bem estabelecidas • Disfibrinogenemia • Hipo ou displasminogenemia • Deficiência do cofator II da heparina • Anormalidades da glicoproteína rica em histidine • Aumento do inibidor do ativador de plasminogênio • Deficiência do ativador do plasminogênio • Trombomodulina anormal
Deficiência de Antitrombina III • Glicoproteína sintetizada no fígado e • meia-vida plasmática de 65 horas. • Herança autossômica dominante, • F:M - igualmente afetados; • Indivíduos heterozigotos - 40 a 70% da atividade funcional da proteína. • Não há descrição de homozigose • Especula-se que a mesma possa ser incompatível com a vida
Fenotipicamente - dois tipos de deficiência AT . • Tipo I - caracterizado pela redução simultânea da atividade funcional e antigênica • alelo mutante não codifica a proteína. • Tipo II - desproporção entre a atividade funcional reduzida, na presença de níveis antigênicos normais • Sugere uma molécula anormal com defeito no centro reativo (fundamental para a ligação com a trombina) • Defeito no sítio de ligação com a heparina ou ambos.
Deficiência de Proteína C • Glicoproteína dependente de vitamina K, • Sintetizada no fígado • Meia vida de 6 a 8 horas. • Herança autossômica dominante • Heterozigotos - níveis variados da proteína • Habitualmente com valores inferiores à 50% da concentração normal • homozigotos - níveis indetectáveis da proteína.
Classificação Fenotípica • Tipo I - redução equivalente da atividade funcional e níveis antigênicos • Tipo II -níveis antigênicos normais e a atividade funcional reduzida • 10% dos casos. • Aproximadamente 160 mutações identificadas no gene da PC. • 60% das mutações tipo I resultam da troca de um aminoácido por outro.
Deficiência de Proteína S • Glicoproteína dependente de vitamina K • Produzida no fígado,células endoteliais, células de Leydig e megacariócitos. • Meia vida de 42 horas • 60% da PS está ligada com a fração C4b do sistema complemento • 40% na forma livre. • Heterozigotos - nível de PS total varia de 30-65% dos valores normais e de PS livre 15-50%. • Homozigotos - níveis inferiores a 5%.
Classificação • Tipo I – redução dos níveis antigênicos da PS total e livre, e concomitante redução da atividade da PS livre. • Tipo II- níveis antigênicos normais de PS total e livre, e redução da atividade funcional. • Tipo III- níveis antigênicos de PS total normais e PS livre com atividade e níveis antigênicos reduzidos.
Resistência à PCa • Principal etiologia de TH. • Adição de PCa purificada ao plasma humano normal • proteólise dos fatores Va e VIIIa e consequente prolongamento do tempo de tromboplastina ativada (ttpa). • resistência à proteína C ativada (RPCa) • Mutação de ponto caracterizada pela transição G→A no nucleotídeo 1691, • resulta na mudança Arg→Gln no codon 506, um dos sítios de ligação com a PCa essa molécula foi denominada fator V Leiden (FVL).
Fator V mutante • inativação deficiente do fator Va com desequilíbrio entre o fator V inativo e ativo → geração maior de trombina e hipercoagulabilidade. • Fenótipo e presente em até 98% dos casos de RPCa. • Heterozigotos - risco para desenvolvimento de trombose venosa 8-10 vezes superior àquele da população geral, • mulheres em uso de ACO ou durante a gravidez – aumento em 30-40 vezes • Prevalência entre indivíduos normais • 2- 10% dos descendentes de caucasóides • menos de 1% entre descendentes de orientais,africanos ou populações indígenas.
Diagnóstico • prolongamento do ttpa pela adição de PCa • pesquisa da mutação Arg506-Gln • amplificação por PCR.
Hiperhomocisteinemia • Resultante de anormalidades funcionais das enzimas do metabolismo da metionina ou da deficiência de cofatores enzimáticos com vitamina B6, B12 ou ácido fólico. • Aumento moderado de homocisteina – variante termolábil da enzima metileno tetrahidrofolato (MTHFR). • Transição 677 C →T no gene da MTHFR • mudança de Ala-Val → redução na ligação com o ác. Fólico → níveis inferiores aos normais de ác.fólico ativo (que apresenta atividade cofatora no metabolismo da homocisteina). • atividade funcional inferior a 50% da forma normal • O risco para o desenvolvimento de trombose é variado entre as distintas populações.
Protrombina mutante • Transição G→A na posição 20.210 na extremidade 3’ do gene • correlação com níveis plasmáticos elevados de protrombina e desenvolvimento de trombose venosa. • Alelo mutante da protrombina identificado em 2,3% da população geral • Elevação para 6% entre indivíduos com pelo menos um episódio de trombose venosa. • prevalência desse alelo mutante varia de 4-7% dos casos de TH. • variante da protrombina - segunda causa mais frequente de TH.
Manifestações Clínicas • Caracterizada pelo desenvolvimento de manifestações tombóticas em indivíduos com idade inferior a 45 anos • TVP e/ou EP observadas na maioria dos casos • Os episódios trombóticos recorrentes ou envolvendo sítios incomuns • veia cava,hepática ou de membros inferiores. • 50% dos casos - história familiar de trombose • defeitos genéticos específicos.
Indivíduos heterozigotos para as deficiências de PC, PS e AT e a presença do fator V Leiden • manifestações clínicas associadas a fatores desencadeantes exógenos • durante o ciclo gravídico puerperal , após cirurgias ou imobilização. • Associação com fatores adquiridos desencadeantes de trombose • mais frequente (62%) em pacientes com fator V Leiden • 30-40% dos casos de deficiência de AT,PC ou PS.
ACO • associado ao aumento do risco de TH • particularmente na presença do fator V Leiden e deficiência de AT. • Episódios trombóticos raramente observados na infância. • Formas homozigóticas para deficiência PC,PS no período neonatal - “purpura fulminans” ( trombose de microcirculação que se manifesta logo após o nascimento).
Diagnóstico • A avaliação global da coagulação pelo tempo de tromboplastina parcial ativado ou tempo de protrombina não permite identificar pacientes com TH. • Necessidade da utilização de métodos específicos para cada fator • custo elevado • limitado a determinados centros. • Seleção dos casos • Baseado no quadro clínico • Reservada para casos onde não há associação com doenças sistêmicas como neoplasias, doenças autoimunes ou síndrome antifosfolípide. • A ausência de história familiar não exclui a possibilidade de TH
Testes laboratoriais específicos Testes diagnósticos iniciais • Hemograma e esfregaço periferico • Tempo de protrombina • TTPA • Tempo de trombina e • Screening para Lupus • Anticardiolipina e anticorpo anti β2glicoproteina • Proteina C ativada • Fibrinogênio • Protrombina mutante (G20210GA) • homocisteína
Testes laboratoriais específicos Testes adicionais específicos • Mutação do fator V leiden • Antitrombina • Atividade Proteína C • Atividade Proteína S • Atividade do Plasminogênio • Teste de trombocitopenia induzido por heparina • Mutação JAK2 por PCR
Testes laboratoriais - quando solicitar História familiar sugestiva Após 1º evento trombótico • Fase aguda: • diminuição da proteína C e S • Aumento do fibrinogênio e fator VIII • Aguardar normalização: em média 6 semanas • Níveis de antitrombina diminuídos em vigência de terapia com heparina • Fatores dependentes da vitamina K diminuídos em vigência de terapia com Warvarin • Teste para fator V de leiden e protrombina mutante não sofrem alterações em vigência de terapia anticoagulante • Resultados alterados • Necessidade de confirmação • Repetir exames - ausência de anticoagulação • Avaliar familiares
Tratamento • Anticoagulação • Ausência de protocolos específicos para crianças • 2 semanas a 3 meses para neonatos • 3 a 6 meses para crianças
Nota DO Editor do site www.paulomargotto.com.br , Dr.Paulo R. Margotto • Consultem Hemorragia intraventricular no recém-nascido a termo, uma condição onde os fatores protrombóticos estão envolvidos.