Download

1 / 21
230 likes | 726 Vues
Mediterranean Anemia-Thalassemia. Kakavoulis Nikolaos Patras Ioannis. What is Thalassaemia ?. Thalassaemia is a group of inherited disorders of hemoglobin synthesis characterized by reduced or absen ce of one or more of the globin chains of adult hemoglobin .

E N D
Mediterranean Anemia-Thalassemia KakavoulisNikolaos Patras Ioannis
What is Thalassaemia ? Thalassaemia is a group of inherited disorders of hemoglobin synthesis characterized by reduced or absenceof one or more of the globin chains of adult hemoglobin . Genetically, it is autosomal recessive blood disease. The name is derived from the Greek words Θάλασσα= Sea" and ”Αίμια= Blood" in reference to anemia of the sea.
Demographics: Thalassemia • Found most frequently in the Mediterranean, Africa, Western and Southeast Asia, India and Burma • 15% of the greek population have the ‘’T’’ gene.
GeneticTypesof Thalassaemia : There are two basic groups of thalassaemia. • Alpha ( )Thalassaemia • Beta ( )Thalassaemia
β Thalassemia • β Thalassemia: deficient/absent beta subunits • Commonly found in Mediterranean, Middle East, Asia, and Africa • Three types: • Minor • Intermedia • Major (Cooley anemia) • May be asymptomatic at birth as HbF functions
Clinical Outcomes of β-Thalassemia β Thalassemia minor (trait) • asymptomatic • microcytosis • minor anemia β Thalassemia intermedia • symptoms similar to Cooley Anemia but less severe β Thalassemia major (Cooley Anemia) • most severe form • moderate to severe anemia • intramedullaryhemolysis (RBC die before full development) • peripheral hemolysis & splenomegaly • skeletal abnormalities (overcompensation by bone marrow) • increased risk of thromboses • pulmonary hypertension & heart failure
Pathophysiology Disturbance of ratio between α & non-α globin chain synthesis then absence or decrease production of one or more globin chains Formation of abnormal Hb structures Ineffective erythropoiesis Excessive RBCs Destruction Iron Overload Extra-medullary hematopoiesis Increased HbF expression
Signs & Symptoms Thalassaemia Minor : Usually no signs or symptoms except for a mild anemia. Thalassaemia Major : 1. Paleness, Jaundice or yellow coloured skin. 2. Growth retardation. 3. Bony abnormalities specially of the facial bones. 4. Enlarged spleen and liver.
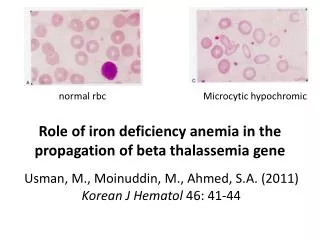
Laboratory Diagnosis • Thalassemia minor: • Haemoglobin : Haemoglobin level is usually normal or mildly reduced. • Peripheral blood film : Hypochromia and Microcytosis (similar to Iron Deficiency Anemia). • MCV< 75 fl, RDW < 14%. • Reticulocyte Count increases • Decrease Osmotic Fragility • Haemoglobinelectrophoresis
Other Special Procedures Globin Chain Testing - determines ratio of globin chains being produced. DNA Analysis - Determine specific defect at molecular DNA level.
Course and treatment of thalassaemia If Untreated • thalassemia Major : Death in first or second decade of life • Intermedia: variable life span • Minor/Minima: Normal life span
Treatment for β Thalassemia • Trait – no treatment required • Intermedia • Major (Cooley anemia) • Regular folate supplementation • RBC transfusion (Splenectomy may decrease need for transfusions) • to maintain [Hgb] ~9-10g/dL • Blood transfusions iron accumulation iron overload • Iron chelators (diferroxamin)
Suggestions for encountering the disease in a more efficient way Raising awareness for more frequent blood donations, since patients with β-thalasseamia require frequent transfusions 8th of May: Thalassemia awareness day.
References http://www.mayoclinic.com/health/thalassemia/DS00905/DSECTION=treatments%2Dand%2Ddrugs http://www.nlm.nih.gov/medlineplus/ency/article/000587.htm http://www.lpch.org/DiseaseHealthInfo/HealthLibrary/hematology/thalbeta.html http://www.nhlbi.nih.gov/health/health-topics/topics/thalassemia/