Download

1 / 14
210 likes | 1.12k Vues
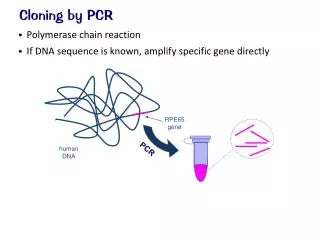
Useful hints and tips for cloning, PCR and site-directed mutagenesis. Kirsten Jensen Division of Molecular Biosciences. Overview. Cloning (of PCR products) Polymerase chain reaction (PCR) Site-directed mutagenesis. MCS. Cloning of a region of interest into a plasmid. Plasmid.

E N D
Useful hints and tips for cloning, PCR and site-directed mutagenesis Kirsten Jensen Division of Molecular Biosciences
Overview • Cloning (of PCR products) • Polymerase chain reaction (PCR) • Site-directed mutagenesis
MCS Cloning of a region of interest into a plasmid Plasmid Region of interest • Choose the right vector (+/- tag, and if tagged, a C- or N-terminal tag). • Blunt end or sticky end cloning. • Check that the enzymes chosen in the MCS for the cloning don’t cut in the region of interest. • Check that there is enough “space” in-between the two enzymes in the MCS. Sticky ends Blunt ends
Primers • Check the reading frame. • C-terminal tag: Add a Kozak consensus sequence (ANNATGG) if it is a C-terminal tag, and again remember to clone the gene in frame with the C-terminal tag. Don’t forget to take the STOP codon out. • Choose enzymes in the MCS to add to the primer that do not cut in the insert. • Primers should be 15-30 nucleotides long and have a GC content of 40-60 %. • The forward and reverse primer should have similar melting temperatures (Tm’s). • Try to avoid primer dimer and hairpin formation. • For sticky end ligation remember to add additional nucleotides to the 5’ side of the restriction site.The restriction site sequences should be followed by 15 bases that are homologous to the template DNA. • The 3’-end of the primer has to end on an C or a G.
Restriction enzymes (NEB) oligo % cleavage sequence 2h 20h BamHI CGGATCCG 10 25 CGGGATCCCG >90 >90 CGCGGATCCGCG >90 >90 EcoRI GGAATTCC >90 >90 CGGAATTCCG >90 >90 CCGGAATTCCGG >90 >90 HindIII CAAGCTTG 0 0 CCAAGCTTGG 0 0 CCCAAGCTTGGG 10 75 NcoI CCCATGGG 0 0 CATGCCATGGCATG 50 75 NdeI GGGTTTCATATGAAACCC 0 0 GGAATTCCATATGGAATTCC 75 >90
The principle of the PCR (Roche) • The purpose of the PCR is to make a huge number of • copies of a gene of interest • 3 steps in a PCR repeated for 30-40 cycles. • Denaturation at 95˚C: the double stranded DNA melts open to single stranded DNA. • Annealing/hybridisation: The primers anneal to the DNA. • Extension 68˚C or 72˚C: The polymerase copies the template adding the dNTPs • from 5’ to 3’.
5’ 3’ 3’ 5’ PCR • 0.1-5 ng plasmid DNA and 0.1-1 ug for genomic DNA • 200 µM dNTPs (final concentration) • 1-1.5 U Taq polymerase and PfuTurbo polymerase 1.25-2.5U • 0.1-1 µM of each primer fwd/rev • 1-4 mM MgCl2 Taq polymerase (MgSO4 for Pfu polymerase) • DMSO up to 10% (has been shown to facilitate DNA separation) • Initial denaturation: 1 Cycle: 1 min. 95˚C is usually enough for plasmid DNA. For genomic DNA it will have to be longer. • 30-40 cycles PCR. • If using primers with restriction sites use a lower annealing temperature for the first 10 cycles of the PCR. • Final extension step: Incubate samples at 68˚C /72˚C for 5-15min to fill in the protruding ends of the PCR products. Taq DNA Polymerase adds extra A nucleotides to the 3'-ends of PCR products. If PCR fragments are cloned into T/A vectors, this step can be prolonged to up to 30min.
Cloning • Digest the vector and the amplified DNA for 2h or O.N. depending on the enzymes used. • Clean up the digested vector and fragment. • Check the concentration of the vector and the fragment on a gel. For Blunt end ligation (ratio vector : fragment; 1pmol : 10pmol) For sticky end ligation (ratio vector : fragment; 1pmol : 3pmol) • Negative control with vector only.
Problems after ligation Colonies on the negative control. • Poor cutting of the vector by the restriction enzymes. De-phosphorylation of the vector (also if using two different enzymes). • Colony screening using PCR. No colonies: • Poor cutting of the PCR product by the restriction enzyme because of inefficient extension by the polymerase. • Poor cutting due to insufficient number of extra nucleotides at the 5’ side of the restriction site. Clone the fragment blunt end and then cut it out. • Blunt end ligation: Use PfuTurbo polymerase.Taq DNA Polymerase adds extra A nucleotides to the 3'-ends of PCR products. Clone the PCR product into a A/T cloning vector or treat it with Klenow and dNTPs and then ligate.
Site-directed mutagenesis Workflow (Stratagene) Gene in plasmid with target site mutation Denature the plasmid and anneal the oligonucleotide primers containing the desired mutation Using the non-strand-displacing action of PfuTurbo polymerase, extend and incorporate the mutagenic primers resulting in nicked circular strands Digest the methylated, nonmutated parental DNA template with Dpn I Transform the circular, nicked dsDNA into super- competent cells After transformation the supercompetent cells repair the nicks in the mutated plasmid
* 3’ 5’ 5’ 3’ * Primer design for site-directed mutagenesis Stratagene: • Both primers must contain the mutation. • The mutation should be in the middle of the primer. • Primers should be 25-45 nucleotides long and have a GC content of at least 40%. • The melting temperature (Tm) should be ≥ 78˚C. • The 3’-end of the primer has to end on an C or a G.
5’ 3’ * 3’ 5’ 5’ * 3’ 3’ 5’ * Primer design for site-directed mutagenesis Invitrogen: • Only one primer contains the mutation. • Both primers should be ~ 30 nucleotides long. • Primers should have an overlapping region at the 5’ end of 15-20 nucleotides. • On the mutagenic primer, there should be at least 10 nucleotides downstream of the mutation. Literature:(Zheng et al. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004 Aug 10;32(14):e115). • Both primers must contain the mutation. • At least 8 non-overlapping bases at the 3’ end. • The 3’-end of the primer has to end on an C or a G.
Site-directed mutagenesis • 5-50 ng/50 µl reactions of dsDNA plasmid DNA from a dam+ E.coli strain • 125-250ng/50 µl reactions of forward and reverse primer (PAGE or HPLC purified) • 200 µM dNTPs • 2 mM MgSO4 • 2.5 U of PfuTurbo polymerase • DMSO up to 10% (has been shown to facilitate DNA separation) Problems: • No product: Don’t continue. • When using DMSO add more PfuTurbo polymerase. • If the oligo anneals several times, extend the oligo and increase the annealing temperature. • Primer dimer and hairpin: Use asymmetric oligos. • Repeats on either site of the mutation: Try shorter oligos and increase the annealing temperature
Papers and useful websites PCR • Barnes WM. PCR amplification of up to 35-kb DNA with high fidelity and high yield from lambda bacteriophage templates. Proc Natl Acad Sci U S A. 1994 Mar 15;91(6):2216-20. • http://www.fermentas.com/ Site-directed mutagenesis • http://www.stratagene.com/ • http://www.invitrogen.com/ • Zheng et al. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004 Aug 10;32(14):e115.