Download

1 / 107
1.12k likes | 1.42k Vues
Mucoviscidosis, celiac disease, lactose intolerance, cow milk allergy. CF. A multisystem disease Autosomal recessive inheritance Cause: mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) chromosome 7 codes for a c-AMP regulated chloride channel. Epidemiology.

E N D
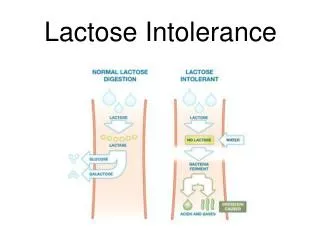
Mucoviscidosis, celiac disease, lactose intolerance, cow milk allergy
CF • A multisystem disease • Autosomal recessive inheritance • Cause: mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) • chromosome 7 • codes for a c-AMP regulated chloride channel
Epidemiology • Most common “life-shortening” recessive genetic disease in Caucasians • 1:3,500 newborns in the US • 1 in 10,500 Native Americans • 1 in 11,500 Hispanics • 1 in 14,000 to 17,000 African Americans • 1 in 25,500 Asians
About 30,000 people affected in United States • >10,000,000 people carriers of mutant CFTR • 80% cases diagnosed by age 3 • Almost 10% diagnosed ≥18 years
PATHOPHISIOLOGY • Mucus-helps clear airway of bacteria • Clearance of mucus depends on • Ciliary function • Mucin secretion • Volume of airway surface liquid (ASL) • Forms periciliary liquid layer (PCL) • Dilutes mucus-facilates entrapment of bacteria and clearance • Optimal volume of ASL regulated by Na+ absorption and Cl- secretion
Airway surface liquid low volume and consequences • Cilia do not beat well when PCL ( periciliary liquid layer)volume is depleted • Mucins are not diluted and cannot be easily swept up the airway • Mucus becomes concentrated • Results in increased adhesion to airway surface • Promotes chronic infection
CONSEQUENCE clinical features of Cystic Fibrosis • Chronic Sino-Pulmonary Disease • Nutritional deficiency/GI abnormality • Obstructive Azoospermia • Electrolyte abnormality • CF in a first degree relative .
General Failure to thrive GI meconium ileus in neonate foul smelling stools, bloating rectal prolapse impaction/obstruction pancreatitis low albumin, low sodium cholelithiasis, cholecystitis cirrhosis, portal hypertension neonatal hyperbilirubinemia fat soluble vitamin deficiency Respiratory clubbing asthma Chronic Obstructive Pulmonary Disease barrel chest productive cough, hemoptysis nasal polyps pneumothorax/mediastinum cor pulmonale, RVH Metabolic dehydration (low Na, low Cl) metabolic alkalosis (esp neonate) DM ( diabetus mellitus) heart stroke GU infertility in males
Endobronchial disease • Hyperinflation • Peribronchial cuffing • Bronchiectasis • Diffuse fibrosis • Atelectasis
Nasal Polyps • Benign lesions in nasal airway • If large enough, can be associated with significant nasal obstruction, drainage, headaches, snoring • Likely associated with chronic inflammation • May need surgical intervention • High recurrence rate
Digital Clubbing • Bulbous swelling at end of fingers • Normal angle between nail and nail bed lost---Schamroth sign • Can be associated with pulmonary disease, cardiac disease, ulcerative colitis, and malignancies
Nutritional deficiency Pancreatic insufficiency • Mucus plugging of glandular ducts • Chloride impermeability affects HCO3- secretion and fluid secretion in pancreatic ducts • Pancreatic enzymes stay in ducts and are activated intraductally • Autolysis of pancreas • Inflammation, calcification, plugging of ducts, fibrosis • Malabsorption • Failure to thrive • Fat soluble vitamin deficiency
GI disease • Intestinal abnormality • Meconium ileus • Distal intestinal obstruction syndrome (DIOS) • Rectal prolapse • Hepatobiliary disease • Focal biliary cirrhosis • Multilobular cirrhosis • Pancreatic endocrine dysfunction • Cystic fibrosis related diabetes
Cystic fibrosis related liver disease • Obstructs biliary ductules • Second leading cause of death in CF • Prevalence 9-37% • Spectrum of disease • increased liver enzymes • biliary cirrhosis • portal hypertension GI tract manifestations (hepatobiliary): Patients may present with a history of jaundice or gastrointestinal tract bleeding.
GI tract manifestations (intestinal) • Neonates: Infants may present with intestinal obstruction at birth and various surgical findings (meconium ileus [7-10% of patients with cystic fibrosis], volvulus, intestinal atresia, perforation, meconium peritonitis). Less commonly, passage of meconium may be delayed (>24-48 h after birth) or cholestatic jaundice may be prolonged. • Infants and children: Patients present with increased frequency of stools, which suggests malabsorption (ie, fat in stools, oil drops in stools), failure to thrive, intussusception (ileocecal), or rectal prolapse.
GI tract manifestations (pancreatic) • Patients with pancreatic insufficiency (PI) have fat-soluble vitamin deficiency and malabsorption of fats, proteins, and carbohydrates (however, malabsorption of carbohydrates is not as severe as that of fats and proteins). Steatorrhea is characterized by frequent, poorly formed, large, bulky, foul-smelling, greasy stools that float in water. Cloth diapers, if used, are difficult to clean. • Patients present with failure to thrive (despite an adequate appetite), flatulence or foul-smelling flatus, recurrent abdominal pain, and abdominal distention. Alternatively, some patients have anorexia without obvious steatorrhea. Many infants have symptoms of gastroesophageal reflux.
Hepatosplenomegaly (fattyliverand portal hypertension) • Rectal prolapse • Dryskin (vitamin A deficiency) • Cheilosis (vitamin B complex deficiency)
Respiratory tract manifestations • Patients present with a chronic or recurrent cough, which can be dry and hacking at the beginning and can produce mucoid (early) and purulent (later) sputum. Prolonged symptoms of bronchiolitisoccur in infants. • Paroxysmal cough followed by vomiting may occur. • Recurrent wheezing, recurrent pneumonia, atypical asthma, pneumothorax, hemoptysis, and digital clubbing are all complications and may be the initial manifestation. • Dyspnea on exertion, history of chest pain, recurrent sinusitis, nasal polyps, and hemoptysis may occur
Physical • Physical signs depend on the degree of involvement of various organs and the progression of disease. • Nose • Rhinitis • Nasal polyps • Pulmonary system • Tachypnea • Respiratory distress with retractions • Wheeze or crackles • Cough (dry or productive of mucoid or purulent sputum) • Increased anteroposterior diameter of chest • Clubbing • Cyanosis • Hyperresonant chest upon percussion: Crackles are heard acutely in associated pneumonitis or bronchitis and chronically with bronchiectasis.
Urogenital tract manifestations • Males are frequently sterile because of the absence of the vas deferens. Undescended testicles or hydrocele may be present. • Fertility is maintained, although possibly decreased, in females. Secondary sexual development is often delayed. • Amenorrhea may occur in patients with severe nutritional or pulmonary involvement.
Other systems • Scoliosis • Kyphosis • Swelling of submandibular gland or parotid gland • Aquagenic wrinkling of the palms (AWP): A recent study reported an association between AWP and cystic fibrosis.Among patients with cystic fibrosis, a greater degree of AWP is observed in patients who are homozygous for the 508delF mutation.
Diagnosis • Elevated serum trypsinogen in neonate • Gene testing • 87 mutation panel (92% sensitivity) • 1300 mutation panel (97 - 99% sensitivity) • GOLD STANDARD: CHLORIDE SWEAT TEST • <40 mEq/L--negative • 40 - 60 mEq/L--equivocal, needs repeat • >60 mEq/L--positive, needs confirmation
Diagnosis of cystic fibrosis • One or more clinical features of CF PLUS • Two CF mutations OR • Two positive quantative pilocarpine iontophoresis sweat chloride values OR • An abnormal nasal transepithelial potential difference value
Diagnosis---Sweat chloride • Technique first described by Gibson and Cooke in 1950s • Chemical that stimulates sweating placed under electrode pad; saline under other electrode pad on arm • Mild electric current is passed between electrodes • Sweat collected
Prenatal screening • American College of Obstetricians and Gynecologists recommended offering patients option of prenatal screening for CF • Carrier testing of 23 most common mutations • Sensitivity of prenatal screening for CF among the white population <78% • lower than that for newborn screening • sensitivity of prenatal testing in racial and ethnic minority populations is lower
Newborn Screening for CF • Goal: diagnose early---evidence that early diagnosis may be associated with better nutritional outcome and chest radiographic scores • Immunoreactive trypsinogen usually first followed by either sweat or DNA testing
False positives adrenal insufficiency nephrogenic DI hypothyroidism mucopolysaccharidosis G6P deficiency hypoproteinemia anemia from poor nutrition False negatives severe malnutrition with edema too little sweat inexperienced tester Differential Diagnosis
Cystic fibrosis---TreatmentMultidisciplinary • Airway Clearance • Infection • Nutrition • Gastrointestinal • Inflammation • Infertility • Social Issues
Treatment • For acute respiratory infections: hospitalization and aminoglycoside, pulmonary toilet • baseline pulmonary therapy • aerosols (bronchodilation) • chest physical therapy • aerosolized antibiotics • systemic steroids or expectorants--not indicated
Treatment: Pulmonary toilet/Airway clearance • Chest physiotherapy • Postural drainage and percussion • P.E.P (positive expiratory pressure valve, Acapella valve, Flutter valve • High frequency chest wall oscillation • Albuterol • Bronchodilation • Increase ciliary efficiency • Dornase alpha/recombinant DNase • Hypertonic Saline by nebulization
Treatment: Chronic infection • Inhaled antibiotics • Inhaled tobramycin in patients with pseudomonas • Sputum cultures • Treatment of pulmonary exacerbation • Pulmonary exacerbation-change in symptoms and signs from baseline (cough, sputum production, lung function, increased crackles on physical exam) • Requires hospitalization for antibiotics IV, as well as increased airway clearance
Treatment: Anti-inflammatory agents • Ibuprofen • Slower decrease in FEV1 annually than placebo group; better weight maintenance • No difference in frequency of hospitalization • Best effect seen in patients less than 13 years of age
Treatment • Nutritional therapy • high fat, high protein diet • pancreatic enzyme replacements • vitamin and mineral supplements • Other • no support for growth hormone • pulmonary transplant--must transplant both lungs simultaneously!
Treatment • Pancreatic Enzymes • These agents aid digestion when the pancreas is malfunctioning. Current pancreatic enzyme preparations are derived from porcine extracts and contain various proportions of lipase, amylase, and protease. Usually, the dose of pancreatic enzymes should not exceed 2000 U/kg/meal of lipase. The novel preparation TheraCLEC-Totalis a highly purified microbiologically-derived enzyme preparation • Pancrelipase (Creon, Pancrease, Ultrase, Viokase) • Enteric-coated pancreatic enzymemicrospherescontainingvariousamounts of lipase, protease, andamylase. Assists in digestion of protein, starch, andfat.500-2000 U of lipase/kg/meal PO; individualize dose to patient; patient's response guides dose; dose of 1-3 cap per meal is sufficient for most patientsAdjust dose according to stool fat and nitrogen content
Tobramycin, inhaled (TOBI) • Formulatedspecifically for inhalation. Chronicintermittentadministration in patientswithP aeruginosainfectionimprovespulmonaryfunctionandnutritional status andreducessymptomaticpulmonaryexacerbation • Aztreonaminhalation (Cayston) • Monobactam antibiotic. Elicitsactivity in vitro against gram-negative aerobic pathogens, includingPseudomonasaeruginosa. Bindstopenicillin-bindingproteins of susceptible bacteria, therebyinhibitingbacterialcellwallsynthesis, resulting in celldeath. Activityisnotdecreased in thepresence of cysticfibrosis lung secretions.Indicatedtoimproverespiratorysymptoms in patientswithcysticfibrosisinfectedwithP aeruginosa.
Gentamicin (Garamycin)3 mg/kg/dose IV q8h • Tobramycin (Nebcin)3 mg/kg/dose IV q8h • Piperacillin (Pipracil)300 mg/kg/d IV divided q6h; not to exceed 24 g/d • Ceftazidime200 mg/kg/d IV divided q6h; not to exceed 6 g/d • Ciprofloxacin (Cipro)20-30 mg/kg/d PO divided q8h • Trimethoprim and sulfamethoxazole (Bactrim, Septra) 8 mg/kg/d (based on TMP component)
Prognosis of CF • Overall trend is improved survival • Female survival worse than male between 2-20 years of age • 35% of patients are older than 18 years of age • Median survival 36.8 years compared to 1930s when life expectancy was about 6 months
ESPGHAN 2012 : Guidelines for the Diagnosis in Children & Adolescents Definition: “CD is an immune-mediated systemic disorder elicited by gluten and related prolamines in genetically susceptible individuals characterised by the presence of a variable combination of gluten dependent clinical manifestations, CD-specific antibodies, HLA-DQ2 or DQ8 haplotypes and enteropathy.”
Coeliac Disease 19882012 • Uncommon Common (1%) • Childhood enteropathy Children & adults Strong genetic predisposition Multi-organ disorder Specific antibody tests
Serology : Gliadin/Reticulin → Tissue Transglutaminase (TG2) → Endomysial (EMA) • Genetics : HLA DQ2/8 • Enteropathy : Variable/patchy
The “Celiac Genes”: HLA DQ2 and DQ8 • Genetic predisposition • Human leukocyte antigen (HLA) alleles DQA1 / DQB1 genes encoding DQ2 and / or DQ8 molecules • Found in 95% of people with CD • 70% concordance in identical twins • Gene test has 100% predictive value to verify when an individual does not have celiac disease.
Epidemiology • May be most common predetermined condition in humans • Found throughout world • Perceived greater incidence in Europe, gluten in diet • Recent screenings found 0.5% to 1% in general population (NIH, 2004; Dube, et al, 2005) • 1/77 Swedish children (Carlsson, et al, 2001) • 1/230 Italian children (Catassi, et al, 1996) • 1/100 5 year old children in Denver (Hoffenberg, et al, 2003) • Ethnic distribution unknown • Only 3% with CD are diagnosed
Pathophysiology • Celiac disease is a multifactorial, autoimmune disorder that occurs in genetically susceptible individuals. • Trigger is an environmental agent-gliadin component of gluten. The enzyme tissue transglutaminase (tTG) has been discovered to be the autoantigen against which the abnormal immune response is directed. • What is gliadin? A glycoprotein present in wheat and other grains such as rye, barley and to some degree, oats. • What is gluten? A composite of the proteins gliadin and glutenin which comprise about 80% of the protein contained in wheat seed.
Normal small intestine Normal villi Small intestine with villous atrophy Small intestine with scalloping
London, year 1938 Classic physical presentation