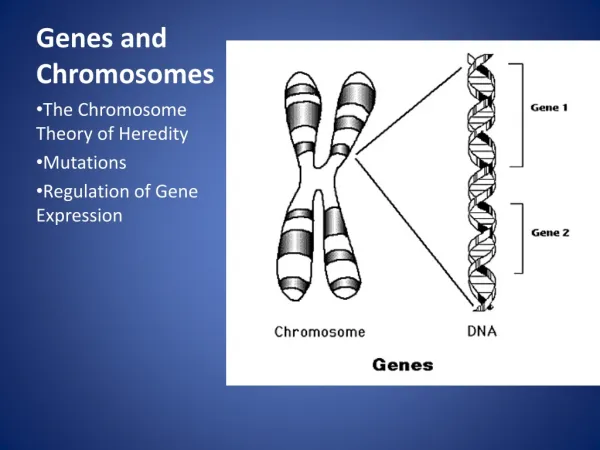
Download
1 / 5
50 likes | 59 Vues
Genes to Trees Daniel Ayres and Adam Bazinet. CMSC858P - Project 2 Proposal. Phylogenetic tree reconstruction. “Genes to Trees”. GenBank. Data collection. Phylogenetic analysis (PAUP, MrBayes, GARLI). Data curation. Multiple sequence alignment (ClustalW, Muscle, MAFFT).

E N D
Genes to Trees Daniel Ayres and Adam Bazinet CMSC858P - Project 2 Proposal
Phylogenetic tree reconstruction “Genes to Trees” GenBank Data collection Phylogenetic analysis (PAUP, MrBayes, GARLI) Data curation Multiple sequence alignment (ClustalW, Muscle, MAFFT) Visual inspection and post-processing
How does it work? • User inputs: • Set of DNA or amino acid sequences • Taxonomic constraints • Homologous sequences obtained from GenBank • Smaller groups eliminated • Multiple alignment of each group made • Uninformative columns removed • “Super-matrix” of all sequences created • Phylogenetics analysis performed • Output: • Phylogenetic tree of closely related organisms Workflow
Is it feasible? • Scripting will be done with Perl • Extensive use of BioPerl libraries • Collection of modules for bioinformatics programming • Accessing sequence data from local and remote databases • Manipulating individual sequences • Searching for similar sequences • Creating and manipulating sequence alignments
Why is this relevant? • Results can serve as a starting point for further analysis • Multiple analyses can be run in parallel • Workflow is modular • A step towards robust, high-throughput phylogenetics