Download
1 / 1
10 likes | 95 Vues
Selecting New BAC clones to be sequenced. Selecting New BAC clones to be sequenced. Assembly Pipeline. Conclusion. Clone Selection Efficiency. BAC sequences assembled by PhredPhrap.

E N D
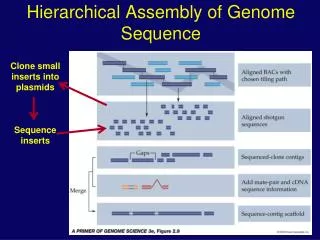
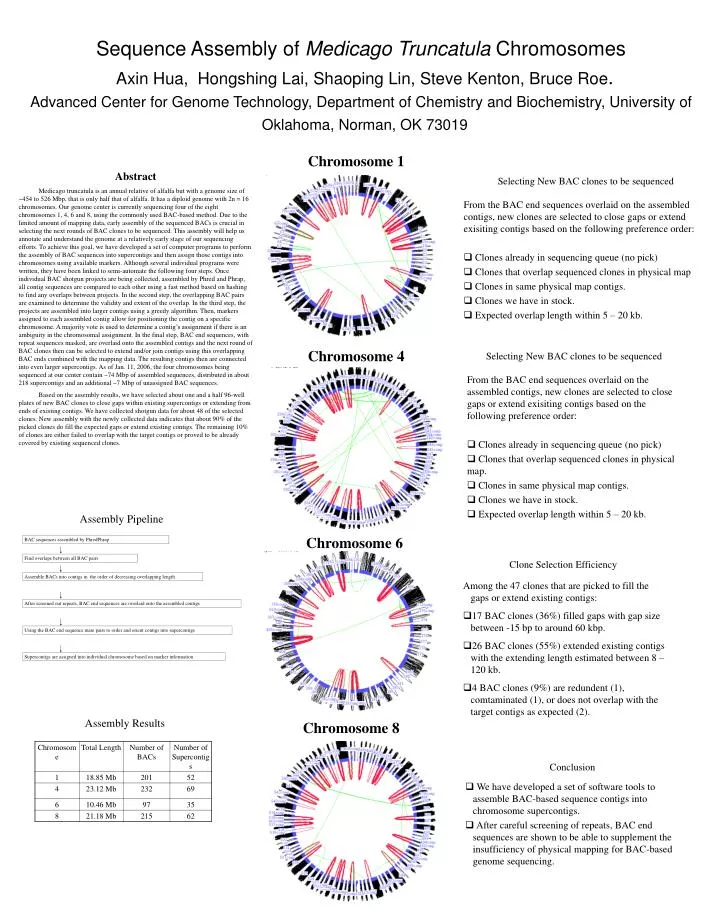
Selecting New BAC clones to be sequenced Selecting New BAC clones to be sequenced Assembly Pipeline Conclusion Clone Selection Efficiency BAC sequences assembled by PhredPhrap • We have developed a set of software tools to assemble BAC-based sequence contigs into chromosome supercontigs. • After careful screening of repeats, BAC end sequences are shown to be able to supplement the insufficiency of physical mapping for BAC-based genome sequencing. • From the BAC end sequences overlaid on the assembled contigs, new clones are selected to close gaps or extend exisiting contigs based on the following preference order: • Clones already in sequencing queue (no pick) • Clones that overlap sequenced clones in physical map. • Clones in same physical map contigs. • Clones we have in stock. • Expected overlap length within 5 – 20 kb. • From the BAC end sequences overlaid on the assembled contigs, new clones are selected to close gaps or extend exisiting contigs based on the following preference order: • Clones already in sequencing queue (no pick) • Clones that overlap sequenced clones in physical map • Clones in same physical map contigs. • Clones we have in stock. • Expected overlap length within 5 – 20 kb. • Among the 47 clones that are picked to fill the gaps or extend existing contigs: • 17 BAC clones (36%) filled gaps with gap size between -15 bp to around 60 kbp. • 26 BAC clones (55%) extended existing contigs with the extending length estimated between 8 – 120 kb. • 4 BAC clones (9%) are redundent (1), comtaminated (1), or does not overlap with the target contigs as expected (2). Find overlaps between all BAC pairs Assemble BACs into contigs in the order of decreasing overlapping length After screened out repeats, BAC end sequences are overlaid onto the assembled contigs Using the BAC end sequence mate pairs to order and orient contigs into supercontigs Supercontigs are assigned into individual chromosome based on marker information Chromosome 8 Sequence Assembly of Medicago Truncatula Chromosomes Axin Hua, Hongshing Lai, Shaoping Lin, Steve Kenton, Bruce Roe. Advanced Center for Genome Technology, Department of Chemistry and Biochemistry, University of Oklahoma, Norman, OK 73019 Chromosome 1 Abstract Medicago truncatula is an annual relative of alfalfa but with a genome size of ~454 to 526 Mbp, that is only half that of alfalfa. It has a diploid genome with 2n = 16 chromosomes. Our genome center is currently sequencing four of the eight chromosomes 1, 4, 6 and 8, using the commonly used BAC-based method. Due to the limited amount of mapping data, early assembly of the sequenced BACs is crucial in selecting the next rounds of BAC clones to be sequenced. This assembly will help us annotate and understand the genome at a relatively early stage of our sequencing efforts. To achieve this goal, we have developed a set of computer programs to perform the assembly of BAC sequences into supercontigs and then assign those contigs into chromosomes using available markers. Although several individual programs were written, they have been linked to semi-automate the following four steps. Once individual BAC shotgun projects are being collected, assembled by Phred and Phrap, all contig sequences are compared to each other using a fast method based on hashing to find any overlaps between projects. In the second step, the overlapping BAC pairs are examined to determine the validity and extent of the overlap. In the third step, the projects are assembled into larger contigs using a greedy algorithm. Then, markers assigned to each assembled contig allow for positioning the contig on a specific chromosome. A majority vote is used to determine a contig’s assignment if there is an ambiguity in the chromosomal assignment. In the final step, BAC end sequences, with repeat sequences masked, are overlaid onto the assembled contigs and the next round of BAC clones then can be selected to extend and/or join contigs using this overlapping BAC ends combined with the mapping data. The resulting contigs then are connected into even larger supercontigs. As of Jan. 11, 2006, the four chromosomes being sequenced at our center contain ~74 Mbp of assembled sequences, distributed in about 218 supercontigs and an additional ~7 Mbp of unassigned BAC sequences. Based on the assembly results, we have selected about one and a half 96-well plates of new BAC clones to close gaps within existing supercontigs or extending from ends of existing contigs. We have collected shotgun data for about 48 of the selected clones. New assembly with the newly collected data indicates that about 90% of the picked clones do fill the expected gaps or extend existing contigs. The remaining 10% of clones are either failed to overlap with the target contigs or proved to be already covered by existing sequenced clones. Chromosome 4 Chromosome 6 Assembly Results