Download

1 / 24
290 likes | 839 Vues
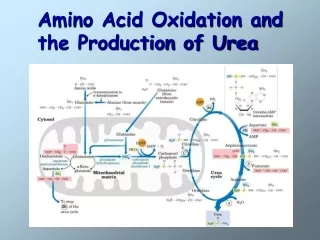
Amino acid oxidation and the production of urea. Nitrogen excretion and the urea cycle Degradation of carbon skeleton of amino acids. Amino acids undergo degradation under circumstances of:. During the normal synthesis and degradation of cellular proteins

E N D
Amino acid oxidation and the production of urea • Nitrogen excretion and the urea cycle • Degradation of carbon skeleton of amino acids
Amino acids undergo degradation under circumstances of: • During the normal synthesis and degradation of cellular proteins • When dietary protein exceeds the body’s needs; amino acids cannot be stored. • During starvation or in diabetes mellitus, cellular proteins are used as fuel.
The first step in the catabolism of amino acids – transamination by aminotransferases (e.g. alanine aminotransferase, aspartate aminotransferase) Amino group donor for biosynthetic pathway Excretion pathway (the coenzyme form of pyridoxine or vitamin B6) PLP is the prothetic group of all aminotransferases
Alanine aminotransferase (ALT; or glutamate-pyruvate transaminase, GPT) Aspartate aminotransferase (AST; or glutamate-oxaloacetate transaminase, GOT) • After a heart attack, a variety of enzymes, including these aminotransferases, leak from the injured heart cells into the bloodstream. Measurements of the blood serum concentration of the two aminotransferases by the SGPT and SGOT test (S for serum), and of another enzyme, creatine kinase, by the SCK test, can provide information about the severity of the damage. • Creatie kinases is the first heart enzyme to appear in the blood after a heart attack; it also disappear quickly from the blood. GOT is the next appear, and GPT follows later. • Liver degeneration caused by carbon tetrachloride, chloroform, or other industrial solvent is accompanied by leakage of GOT and GPT from injured hepaocytes into blood.
Excretion pathway Oxidative deamination (Extrahepatic tissues) (Liver mitochondria) Fix NH4+ in extrahepatic tissues Release NH4+ in liver (Blood) Biosynthesis of amino acids (all tissues) Release NH4+ in liver & kidney Kidney Urine Liver NH4+ (glutamate dehydrogenase) Carbon skeleton
Alanine transports ammonia from muscles to the liver via the glucose-alanine cycle • Low [NH4+]blood • when breakdown muscle protein for energy (An ammonia carrier in blood)
Ammonia is toxic to animals • Depletion of a-ketoglutarate to glutmate by glutamate dehydrogenase and conversion of glutamate to glutamine by glutamine synthetase to consume the cytosol of excess ammonia, limiting the availability of a-ketoglutarate for the citric acid cycle. The glutamine synthetase reaction depletes ATP. Overall, toxic concentration of NH4+ may interfere with the very high levels of ATP production required to maintain brain function. • Depletion of glutamate in the glutamine synthetase reaction may additional effects on the brain. Glutamate and its derivative, -aminobutyrate (GABA), are important neurotransmitters. • The sensitivity of the brain to ammonia may reflect a depletion of neurotransmitters as well as changes in cellular ATP metabolism.
The urea cycle liver mitochondria→.kidney→ urine 1.Ornithine transcarbamoylase 2. Argininosuccinate synthetase 3. Argininosuccinate lyase 4. arginase (Rate-limiting step)
Carbamoyl phosphate synthetase I HCO3 HCO3 HCO3 Carbamate Urea Carbamate Arginine Ornithine Argininosuccinate synthetase mechanism
The overall equation of the urea cycle is 2 NH4+ + HCO3- + 3 ATP4- + H2O → Urea + 2 ADP3- + 4 Pi2- + AMP2- + 5 H+ - 2.5 ATP
Several enzyme cofactors play important roles in amino acid catabolism Three enzyme cofactors important in one-carbon transfer reaction
Synthesis of methionine and S-adenosylmethionine in an activated-methyl cycle
Methionine synthase present in bacteria and mammals uses either N5-methyl-tetrahydrofolate or methylcobalamin derived from coenzyme B12. • In cases of vitamin B12 deficiency, some symptoms can be alleviated by the administration not only of vitamin B12 but of folate. • The methyl group of methylcobalamin is derived from N5-methyl-tetrahydrofolate . Because the reaction converting the N5, N10-methylene form to the N5-methyl form of tetrahydrofolate is irreversible, if coenzyme B12 is not available for the synthesis of methylcobalamin, then no acceptor is available for the methyl group of N5-methyltetrahydrofolate and metabolic folates become trapped in the N5-methyl form.
Phenylketonuria (PKU) • A genetic defect in phenylalanine hydroxylase, the first enzyme in the catalbolic pathway for phenylalanine. • The most common cause of elevated levels of phenylalanine (hyperphenylalanine). • Phenylalanine hydroxylase is one of a general classes of enzymes called mixed-function oxidases.
Role of tetrahydrobiopterin in the phenylalanine hydroxylase reaction. Note that NADH is required to restore the reduced form of the coenzyme.
Branched-chain amino acids are not degraded in the liver • Although much of the catabolism of amino acids takes place in the liver, the three amino acids with branched side chains (leucine, isoleucine, and valine) are oxidized as fuels primarily in muscle, adipose, kidney, and brain tissue. • These extrahepatic tissues contain an aminotransferase, absent in liver, that acts on all three branched-chain amino acids to produce the corresponding a-keto acids. • The branched-chain a-keto acid dehydrogenase complex then catalyzes oxidative decarboxylation of all three a-keto acids, in each case releasing the carboxyl group as CO2 and producing the acyl-CoA derivative.
Catabolic pathways for the three branched-chain amino acids: valine, isoleucine, and leucine
Maple syrup urine disease (MUSD) • The characteristic odor imparted to the urine by the a-keto acids, results from a defective branched-chain a-ketoacid dehydrogenase complex. • Untreated, the disease results in abnormal development of the brain, mental retardation, and death in early infancy. • Treatment entails rigid control of the diet, limiting the intake of valine, isoleucine, and leucine to the minimum required to permit normal growth.