Download
1 / 1
20 likes | 190 Vues
Toward the Identification of a Gene Expression Framework in Different Types of Tissues and Organisms. Saulo Augusto de Paula Pinto 1, 2 { saulo@pucminas.br }. J. Miguel Ortega 1 { miguel@icb.ufmg.br }. 2 Instituto de Informática PUC MINAS BARREIRO. 1 Laboratório de Biodados

E N D
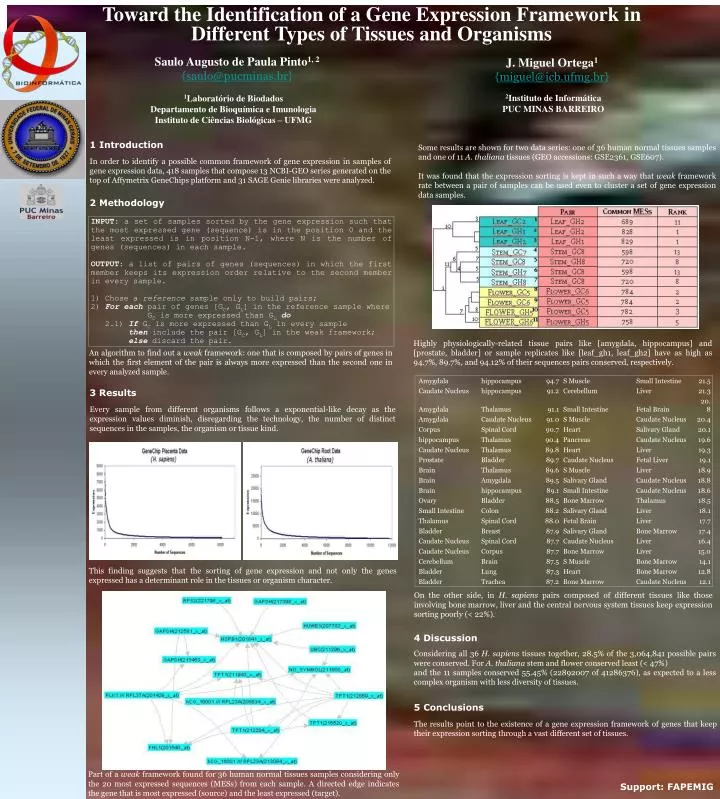
Toward the Identification of a Gene Expression Framework in Different Types of Tissues and Organisms Saulo Augusto de Paula Pinto1, 2 {saulo@pucminas.br} J. Miguel Ortega1 {miguel@icb.ufmg.br} 2Instituto de Informática PUC MINAS BARREIRO 1Laboratório de Biodados Departamento de Bioquímica e Imunologia Instituto de Ciências Biológicas – UFMG 1 Introduction In order to identify a possible common framework of gene expression in samples of gene expression data, 418 samples that compose 13 NCBI-GEO series generated on the top of Affymetrix GeneChips platform and 31 SAGE Genie libraries were analyzed. 2 Methodology Some results are shown for two data series: one of 36 human normal tissues samples and one of 11 A. thaliana tissues (GEO accessions: GSE2361, GSE607). It was found that the expression sorting is kept in such a way that weak framework rate between a pair of samples can be used even to cluster a set of gene expression data samples. Highly physiologically-related tissue pairs like [amygdala, hippocampus] and [prostate, bladder] or sample replicates like [leaf_gh1, leaf_gh2] have as high as 94.7%, 89.7%, and 94.12% of their sequences pairs conserved, respectively. An algorithm to find out a weak framework: one that is composed by pairs of genes in which the first element of the pair is always more expressed than the second one in every analyzed sample. 3 Results Every sample from different organisms follows a exponential-like decay as the expression values diminish, disregarding the technology, the number of distinct sequences in the samples, the organism or tissue kind. This finding suggests that the sorting of gene expression and not only the genes expressed has a determinant role in the tissues or organism character. On the other side, in H. sapiens pairs composed of different tissues like those involving bone marrow, liver and the central nervous system tissues keep expression sorting poorly (< 22%). 4 Discussion Considering all 36 H. sapiens tissues together, 28.5% of the 3,064,841 possible pairs were conserved. For A. thaliana stem and flower conserved least (< 47%) and the 11 samples conserved 55.45% (22892007 of 41286376), as expected to a less complex organism with less diversity of tissues. 5 Conclusions The results point to the existence of a gene expression framework of genes that keep their expression sorting through a vast different set of tissues. Part of a weak framework found for 36 human normal tissues samples considering only the 20 most expressed sequences (MESs) from each sample. A directed edge indicates the gene that is most expressed (source) and the least expressed (target). Support: FAPEMIG











