Download

1 / 26
270 likes | 406 Vues
Local Alignment and BLAST. Three key questions. Query? Purpose? Database?. BLAST – the way it used to look. >gi|77630012|ref|ZP_00792598.1| COG0442: Prolyl-tRNA synthetase [Yersinia pseudotuberculosis IP 31758] Length=572

E N D
Three key questions • Query? • Purpose? • Database?
>gi|77630012|ref|ZP_00792598.1| COG0442: Prolyl-tRNA synthetase [Yersinia pseudotuberculosis IP 31758] Length=572 Score = 1013 bits (2619), Expect = 0.0, Method: Composition-based stats. Identities = 498/572 (87%), Positives = 537/572 (93%), Gaps = 0/572 (0%) Query 1 MRTSQYMLSTLKETPADAEVISHQLMLRAGMIRKLASGLYTWLPTGLRVLRKVENIVREE 60 MRTSQY+LST KETPADAEVISHQLMLRAGMIRKLASGLYTWLPTG+RVL+KVENIVREE Sbjct 1 MRTSQYLLSTQKETPADAEVISHQLMLRAGMIRKLASGLYTWLPTGVRVLKKVENIVREE 60 Query 61 MNNAGAIEVSMPVVQPADLWVESGRWDQYGPELLRFVDRGERPFVLGPTHEEVITDLIRN 120 MNNAGAIEVSMPVVQPADLW ESGRW+QYGPELLRFVDRGERPFVLGPTHEEVITDLIR Sbjct 61 MNNAGAIEVSMPVVQPADLWQESGRWEQYGPELLRFVDRGERPFVLGPTHEEVITDLIRG 120 Query 121 EVSSYKQLPLNFFQIQTKFRDEVRPRFGVMRSREFLMKDAYSFHTSQESLQATYDTMYAA 180 E++SYKQLPLNFFQIQTKFRDEVRPRFGVMR+REFLMKDAYSFHT+QESLQ TYD MY A Sbjct 121 EINSYKQLPLNFFQIQTKFRDEVRPRFGVMRAREFLMKDAYSFHTTQESLQETYDAMYTA 180 …………………………. Query 481 MNMHKSFRVKEVAEDIYQQLRAKGIEVLLDDRKERPGVMFADMELIGVPHTIVIGDRNLD 540 MNMHKSFRVKE+AE++Y LR+ GI+V+LDDRKERPGVMFADMELIGVPH IVIGDRNLD Sbjct 481 MNMHKSFRVKELAEELYTTLRSHGIDVILDDRKERPGVMFADMELIGVPHNIVIGDRNLD 540 Query 541 SEEIEYKNRRVGEKQMIKTSEIIDFLLANIIR 572 SEE+EYKNRRVGEKQMIKTSEI++FLL+ I R Sbjct 541 SEEVEYKNRRVGEKQMIKTSEIVEFLLSQIKR 572
Global Alignment vs. Local Alignment • Global Methods find the best alignment of both sequences in their entirety • Local Methods find the best alignable subsections of both sequences
Sequence Similarity Searches using BLAST BLAST: Basic Local Alignment Search Tool Altschul, S.F., Gish, W., Miller, W., Myers, E.W. & Lipman, D.J. J Mol Biol. 1990 Oct 5;215(3):403-10. Statistical basis: Karlin, S., and Altschul, S. F. (1990) ``Method for assessing the statistical significance of molecular sequence features by using general scoring schemes,'' Proceedings of the National Academy of Science, USA 87, 2264-2268.
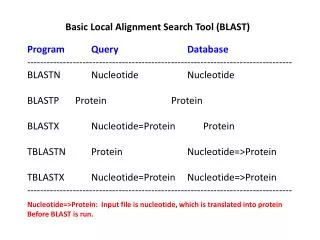
Comparing a Genome to Other Genes and Genomes BLAST = Basic Local Alignment Search Tool BLASTN DNA sequence vs. DNA sequence db BLASTP protein sequence vs. protein sequence db BLASTX DNA sequence translated in 6 reading frames vs. protein sequence db tBLASTX DNA sequence translated in 6 reading frames vs. DNA sequence db translated in 6 frames PSI-BLAST Iterative Search
Comparing a Genome to Other Genes and Genomes BLAST = Basic Local Alignment Search Tool • Find a potential match in the database by finding a little seed (or seeds) of a match • 2. Extend that seed and score the resulting alignment based on co-occurance of amino acids (nucleotides) in “known” alignments • Determine whether the possible alignment looks better than you might expect by chance alone. • 4. Decide whether the match tells you anything about biology.
Find a potential match in the database by finding a little seed (or seeds) of a match db query Your query is small relative to the universe of known sequences
N Y A L L P W M T A Y E N V Y L A V D V F Q N E L L P WR N V Q D N V A F G 2. Extend the seed and score the resulting alignment based on co-occurance of amino acids (nucleotides) in “known” alignments
Alignment Methods – Dynamic Programming • Needleman-Wunsch (global) and Smith-Waterman (local) use dynamic programming • Guaranteed to find an optimal alignment given a particular scoring function • Computationally intensive
Dynamic Programming • One possible simple scoring scheme: • Si,j = 1 if the residue at position i of sequence #1 is the same as the residue at position j of sequence #2 (match score); otherwise • Si,j = 0 (mismatch score) • w = 0 (gap penalty)
Dynamic Programming Three steps: 1) Initialize 2) Fill Matrix Mi,j = MAXIMUM [Mi-1, j-1 + Si,j (match/mismatch in the diagonal), Mi,j-1 + w (gap in sequence #1), Mi-1,j + w (gap in sequence #2)]
Dynamic Programming 3) Traceback G A A T T C A G T T A G G A - T C - G - - A Score = 1+0+1+0+1+1+0+1+0+0+1 = 6
How does BLASTP score an alignment? Substitution Matrix based on co-occurrence in related proteins BLOSUM = BLOcks Substitution Matrix Identify gap-free protein alignments in the BLOCKS database. BLOSUM# corresponds to % identity for inclusion Count co-occurrence of Amino Acids in alignment Calculate log-odds ratio: Log (observed frequency/expected frequency)
How does BLASTP score an alignment? Substitution Matrix based on co-occurance in related proteins 62 means that contributions from proteins more than 62% identical are weighted to sum to one. Other matrices are available for comparisons of more or less divergent proteins.
How does BLASTP score an alignment? Walk through the alignment and add up the score Query: AFGECDA AF C+A Sbjct: AFAFCEA 4+6+0+(-3)+9+2+4 = 22 Normalize bit score
Statistics of BLAST when no gaps are allowed • The number of matches (E) expected to occur with a score as good as S just by random chance, when you search a sequence the size of your query against a database as large as the one you chose (m and n), tends to follow an Extreme Values Distribution (K and lambda). • Simulation is used to estimate K and lambda for gapped BLAST
How good is your BLAST hit? • The number of matches (E) expected to occur with a score as good as S just by random chance >gi|77630012|ref|ZP_00792598.1| COG0442: Prolyl-tRNA synthetase [Yersinia pseudotuberculosis IP 31758] Length=572 Score = 1013 bits (2619), Expect = 0.0, Method: Composition-based stats. Identities = 498/572 (87%), Positives = 537/572 (93%), Gaps = 0/572 (0%) Query 1 MRTSQYMLSTLKETPADAEVISHQLMLRAGMIRKLASGLYTWLPTGLRVLRKVENIVREE 60 MRTSQY+LST KETPADAEVISHQLMLRAGMIRKLASGLYTWLPTG+RVL+KVENIVREE Sbjct 1 MRTSQYLLSTQKETPADAEVISHQLMLRAGMIRKLASGLYTWLPTGVRVLKKVENIVREE 60
Search one protein against a given database and most of the E values are zero
Search one protein against a given database and most of the E values are zero Search the protein encoded by the gene next to it in the genome against the same database and all the E values are much higher.
Search the same protein against two different databases and the E value is different for the same hit.
So,what’s a good match? E-values… 0.0 is a perfect score Really good matches have really small E-values, like e-107 Matches can still be real with moderate E-values like e-05 Sometimes matches with higher E-values are still real. You should also have some expectation of what level of match is typical for the type of comparison. For example: if you are querying with the E. coli O157:H7 proteins against a database of E. coli K-12 proteins, and most orthologous proteins have matches on the order of e-107, then a match that scores e-05 is probably not an orthologous pair.
Other characteristics of a good match: Amount of the sequences in the alignment A match that includes >90% of both sequences is great Divergent matches include blocks of higher identity Conserved motifs can be indicative of conserved function All equally good matches are to proteins with the same function ***The function of at least one of the best hits was experimentally determined.***