Download

1 / 56
620 likes | 1.2k Vues
. CC: Nausea/ vomiting x 6 mos, CP x 4 days, worse with deep inspiration; ?All my joints hurting/swollen." x 48 hrs HPI: Pt is a 45 YO male with PMHx HCV, Barrett's esophagus, admitted with 4 day hx of pleuritic CP, nausea and vomiting x 6 months, 80 lb weight loss over 6 mos. Pt attributes his w

E N D
1. Systemic Sclerosis Heidi Roppelt, MD
Assistant Professor of Medicine
Associate Program Director Division of Rheumatology
2. CC: Nausea/ vomiting x 6 mos, CP x 4 days, worse with deep inspiration; �All my joints hurting/swollen.� x 48 hrs
HPI: Pt is a 45 YO male with PMHx HCV, Barrett�s esophagus, admitted with 4 day hx of pleuritic CP, nausea and vomiting x 6 months, 80 lb weight loss over 6 mos. Pt attributes his weight loss to difficulty swallowing solids, and persistent nausea.
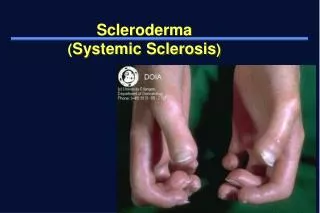
Pt states he has had joint pain and swelling in both hands, both knees, and both ankles off and on for > 1 year, with �hard white bumps.� on his fingertips which he removes with a razor blade. Also complains of red rash anterior chest, raynaud�s.
3. Pt also complains of �My skin is shrinking- I feel like a lizard.� He has noted thickening of the skin on his face, chest arms, hands, which has been ongoing for > 1-2 yrs.
4. PE: BP= 108/69 HR= 78 RR= 12
HEENT: skin thickening over entire face, with telangiectasias
Decrease mouth opening
SKIN: diffuse skin thickening over hands extending to upper arms, anterior chest, with telangiectasias, and areas of hypopigmentation
PULM: dray crackles at bases bilat with occasional diffuse expiratory wheezes
GI: abdomen slightly distended; no ascites appreciated, no organomegaly
Extremities: sclerodactyly, dupytrene�s contractures, ulceration of the fingertips, bilat knee effusions; wrist, MCP, PIP swelling, warmth, pain, redness
9. Labs and studies:
10. CT chest:ground glass opacities consistent with ILD; fluid filled dilated mid-distal esophagus with enhancing thickened wall
ECHOCARDIOGRAM:nl EF, nl estimated PA pressures, moderate anterior and posterior pericardial effusions
PFT: restrictive pattern with low diffusion capacity
11. Forearm/ hand x-rays: no calcinosis; presence of acro-osteolysis R 3rd digit, L 5th digit.
Endoscopy: erosive esophagitis with areas of desquamation and ulceration
12. 45 YO male with serositis, polyarthritis, diffuse skin thickening, nausea/ vomiting, weight loss, +ANA, telangiectasias, esophageal dysmotility, sclerodactyly, abnormal PFT�s (restrictive pattern)
This patient�s diagnosis is?
A- systemic sclerosis- limited
B-systemic sclerosis diffuse
C-CREST
D-sclermyxedema
13. B- systemic sclerosis- diffuse
14. Systemic sclerosis: pathogenesis Abnormalities in three types of cells: fibroblasts, endothelial cells, cells of the immune system (T and B)
Abnormalities in the above cells result in triad of pathological changes:
progressive cutaneous and visceral fibrosis
obliteration of the lumen of small arteries and arterioles
Humoral and cellular immunologic abnormalities (autoantibodies, chronic mononuclear cell infiltration, dysregulation of lymphokine, and growth factor production) The pathogenesis of systemic sclerosis is extremely complex. At present, no single unifying hypothesis explains all aspects of its pathogenesis. However, fundamental abnormalities in at least 3 types of cells are intimately involved in the development of the clinical and pathologic manifestations of the disease: 1) fibroblasts (see Glossary); 2) endothelial cells; and 3) cells of the immune system, particularly T and B lymphocytes. The profound functional alterations in these cells result in the characteristic triad of pathologic changes in systemic sclerosis: severe and often progressive cutaneous and visceral fibrosis; obliteration of the lumen of small arteries and arterioles; and humoral and cellular immunologic abnormalities, which include the production of numerous autoantibodies (some very highly specific for the disease), the chronic mononuclear-cell infiltration of affected tissues, and the dysregulation of lymphokine and growth factor production (see Glossary). These dysfunctional cellular processes and their resulting effects on the affected tissues are illustrated in Figure 1. At present, it is not clear which of these alterations is of primary importance or how they interrelate to cause the progressive fibrotic process in systemic sclerosis. However, a crucial component in the pathogenesis of the disorder is the persistent and unregulated activation of genes encoding various collagens and other extracellular matrix proteins in systemic sclerosis fibroblasts. This is the most important difference between normal fibroblasts, which promote normal wound healing, and systemic sclerosis fibroblasts, which demonstrate an uncontrolled production and tissue deposition of collagen resulting in pathologic organ fibrosis. This paper reviews some of the numerous components of the complex puzzle of systemic sclerosis pathogenesis, as illustrated in Figure 1, and attempts to form hypotheses that may provide frameworks for the development of novel and effective therapies. Because we were not able to provide a comprehensive discussion of the abundant literature published, we refer readers to recent reviews for a more complete and detailed description.
The pathogenesis of systemic sclerosis is extremely complex. At present, no single unifying hypothesis explains all aspects of its pathogenesis. However, fundamental abnormalities in at least 3 types of cells are intimately involved in the development of the clinical and pathologic manifestations of the disease: 1) fibroblasts (see Glossary); 2) endothelial cells; and 3) cells of the immune system, particularly T and B lymphocytes. The profound functional alterations in these cells result in the characteristic triad of pathologic changes in systemic sclerosis: severe and often progressive cutaneous and visceral fibrosis; obliteration of the lumen of small arteries and arterioles; and humoral and cellular immunologic abnormalities, which include the production of numerous autoantibodies (some very highly specific for the disease), the chronic mononuclear-cell infiltration of affected tissues, and the dysregulation of lymphokine and growth factor production (see Glossary). These dysfunctional cellular processes and their resulting effects on the affected tissues are illustrated in Figure 1. At present, it is not clear which of these alterations is of primary importance or how they interrelate to cause the progressive fibrotic process in systemic sclerosis. However, a crucial component in the pathogenesis of the disorder is the persistent and unregulated activation of genes encoding various collagens and other extracellular matrix proteins in systemic sclerosis fibroblasts. This is the most important difference between normal fibroblasts, which promote normal wound healing, and systemic sclerosis fibroblasts, which demonstrate an uncontrolled production and tissue deposition of collagen resulting in pathologic organ fibrosis. This paper reviews some of the numerous components of the complex puzzle of systemic sclerosis pathogenesis, as illustrated in Figure 1, and attempts to form hypotheses that may provide frameworks for the development of novel and effective therapies. Because we were not able to provide a comprehensive discussion of the abundant literature published, we refer readers to recent reviews for a more complete and detailed description.
15. Systemic sclerosis: pathogenesis Persistent and unregulated activation of genes, and abnl regulation of transcription of genes encoding collagens, and extracellular matrix proteins
Alterations in the production of cytokines, and growth factors, which modulate the function of fibroblasts, etc.
TGF- beta is intimately involved in the fibrosis The pathogenesis of systemic sclerosis is extremely complex. At present, no single unifying hypothesis explains all aspects of its pathogenesis. However, fundamental abnormalities in at least 3 types of cells are intimately involved in the development of the clinical and pathologic manifestations of the disease: 1) fibroblasts (see Glossary); 2) endothelial cells; and 3) cells of the immune system, particularly T and B lymphocytes. The profound functional alterations in these cells result in the characteristic triad of pathologic changes in systemic sclerosis: severe and often progressive cutaneous and visceral fibrosis; obliteration of the lumen of small arteries and arterioles; and humoral and cellular immunologic abnormalities, which include the production of numerous autoantibodies (some very highly specific for the disease), the chronic mononuclear-cell infiltration of affected tissues, and the dysregulation of lymphokine and growth factor production (see Glossary). These dysfunctional cellular processes and their resulting effects on the affected tissues are illustrated in Figure 1. At present, it is not clear which of these alterations is of primary importance or how they interrelate to cause the progressive fibrotic process in systemic sclerosis. However, a crucial component in the pathogenesis of the disorder is the persistent and unregulated activation of genes encoding various collagens and other extracellular matrix proteins in systemic sclerosis fibroblasts. This is the most important difference between normal fibroblasts, which promote normal wound healing, and systemic sclerosis fibroblasts, which demonstrate an uncontrolled production and tissue deposition of collagen resulting in pathologic organ fibrosis. This paper reviews some of the numerous components of the complex puzzle of systemic sclerosis pathogenesis, as illustrated in Figure 1, and attempts to form hypotheses that may provide frameworks for the development of novel and effective therapies. Because we were not able to provide a comprehensive discussion of the abundant literature published, we refer readers to recent reviews for a more complete and detailed description.
The pathogenesis of systemic sclerosis is extremely complex. At present, no single unifying hypothesis explains all aspects of its pathogenesis. However, fundamental abnormalities in at least 3 types of cells are intimately involved in the development of the clinical and pathologic manifestations of the disease: 1) fibroblasts (see Glossary); 2) endothelial cells; and 3) cells of the immune system, particularly T and B lymphocytes. The profound functional alterations in these cells result in the characteristic triad of pathologic changes in systemic sclerosis: severe and often progressive cutaneous and visceral fibrosis; obliteration of the lumen of small arteries and arterioles; and humoral and cellular immunologic abnormalities, which include the production of numerous autoantibodies (some very highly specific for the disease), the chronic mononuclear-cell infiltration of affected tissues, and the dysregulation of lymphokine and growth factor production (see Glossary). These dysfunctional cellular processes and their resulting effects on the affected tissues are illustrated in Figure 1. At present, it is not clear which of these alterations is of primary importance or how they interrelate to cause the progressive fibrotic process in systemic sclerosis. However, a crucial component in the pathogenesis of the disorder is the persistent and unregulated activation of genes encoding various collagens and other extracellular matrix proteins in systemic sclerosis fibroblasts. This is the most important difference between normal fibroblasts, which promote normal wound healing, and systemic sclerosis fibroblasts, which demonstrate an uncontrolled production and tissue deposition of collagen resulting in pathologic organ fibrosis. This paper reviews some of the numerous components of the complex puzzle of systemic sclerosis pathogenesis, as illustrated in Figure 1, and attempts to form hypotheses that may provide frameworks for the development of novel and effective therapies. Because we were not able to provide a comprehensive discussion of the abundant literature published, we refer readers to recent reviews for a more complete and detailed description.
16. The pathogenesis of systemic sclerosis is extremely complex. At present, no single unifying hypothesis explains all aspects of its pathogenesis. However, fundamental abnormalities in at least 3 types of cells are intimately involved in the development of the clinical and pathologic manifestations of the disease: 1) fibroblasts (see Glossary); 2) endothelial cells; and 3) cells of the immune system, particularly T and B lymphocytes. The profound functional alterations in these cells result in the characteristic triad of pathologic changes in systemic sclerosis: severe and often progressive cutaneous and visceral fibrosis; obliteration of the lumen of small arteries and arterioles; and humoral and cellular immunologic abnormalities, which include the production of numerous autoantibodies (some very highly specific for the disease), the chronic mononuclear-cell infiltration of affected tissues, and the dysregulation of lymphokine and growth factor production (see Glossary). These dysfunctional cellular processes and their resulting effects on the affected tissues are illustrated in Figure 1. At present, it is not clear which of these alterations is of primary importance or how they interrelate to cause the progressive fibrotic process in systemic sclerosis. However, a crucial component in the pathogenesis of the disorder is the persistent and unregulated activation of genes encoding various collagens and other extracellular matrix proteins in systemic sclerosis fibroblasts. This is the most important difference between normal fibroblasts, which promote normal wound healing, and systemic sclerosis fibroblasts, which demonstrate an uncontrolled production and tissue deposition of collagen resulting in pathologic organ fibrosis. This paper reviews some of the numerous components of the complex puzzle of systemic sclerosis pathogenesis, as illustrated in Figure 1, and attempts to form hypotheses that may provide frameworks for the development of novel and effective therapies. Because we were not able to provide a comprehensive discussion of the abundant literature published, we refer readers to recent reviews for a more complete and detailed description The pathogenesis of systemic sclerosis is extremely complex. At present, no single unifying hypothesis explains all aspects of its pathogenesis. However, fundamental abnormalities in at least 3 types of cells are intimately involved in the development of the clinical and pathologic manifestations of the disease: 1) fibroblasts (see Glossary); 2) endothelial cells; and 3) cells of the immune system, particularly T and B lymphocytes. The profound functional alterations in these cells result in the characteristic triad of pathologic changes in systemic sclerosis: severe and often progressive cutaneous and visceral fibrosis; obliteration of the lumen of small arteries and arterioles; and humoral and cellular immunologic abnormalities, which include the production of numerous autoantibodies (some very highly specific for the disease), the chronic mononuclear-cell infiltration of affected tissues, and the dysregulation of lymphokine and growth factor production (see Glossary). These dysfunctional cellular processes and their resulting effects on the affected tissues are illustrated in Figure 1. At present, it is not clear which of these alterations is of primary importance or how they interrelate to cause the progressive fibrotic process in systemic sclerosis. However, a crucial component in the pathogenesis of the disorder is the persistent and unregulated activation of genes encoding various collagens and other extracellular matrix proteins in systemic sclerosis fibroblasts. This is the most important difference between normal fibroblasts, which promote normal wound healing, and systemic sclerosis fibroblasts, which demonstrate an uncontrolled production and tissue deposition of collagen resulting in pathologic organ fibrosis. This paper reviews some of the numerous components of the complex puzzle of systemic sclerosis pathogenesis, as illustrated in Figure 1, and attempts to form hypotheses that may provide frameworks for the development of novel and effective therapies. Because we were not able to provide a comprehensive discussion of the abundant literature published, we refer readers to recent reviews for a more complete and detailed description
17. Systemic sclerosis: pathogenesis Possible Causative Agents
strong evidence that genetic factors play an important role; however, environmental agents may be more influential
One study reported a remarkably low concordance in the development of systemic sclerosis among homozygous twins
Many infectious, chemical, and physical agents have been postulated as being involved in the cause of the disease
molecular mimicry
herpesviruses, retroviruses, and human cytomegalovirus infections as possible causative agents The cause of systemic sclerosis has remained elusive despite intense investigations. Although the disease is not inherited in a classical Mendelian pattern, there is strong evidence that genetic factors contribute to its development and clinical manifestations, as discussed in more detail shortly. However, it has become apparent that environmental agents play a crucial and more important role than genetic influences. One study reported a remarkably low concordance in the development of systemic sclerosis among homozygous twins, indicating that the heritability component of the disease was very low and that the most important factor was of an environmental or acquired origin (17). Many infectious, chemical, and physical agents have been postulated as being involved in the cause of the disease. The hypothesis that infectious agents may cause systemic sclerosis has been studied extensively. Some researchers have suggested that the production of specific autoantibodies in systemic sclerosis is the result of an antigen-driven response caused by "molecular mimicry." The concept of "molecular mimicry" proposes that antibodies against self-antigens are produced because these antigens contain epitopes (see Glossary) that share structural similarities with viral or bacterial proteins. In the immunopathogenesis of systemic sclerosis, herpesviruses, retroviruses, and human cytomegalovirus infections, among others, have been suggested as possible causative agents. Evidence supporting the role of retroviruses includes the demonstration of sequence homologies (see Glossary) between certain retroviral proteins and the topoisomerase I antigen (see Glossary), which is the target of anti�Scl-70 antibodies in patients with systemic sclerosis (18). In addition, it has been shown that the induced expression of retroviral proteins in normal human dermal fibroblasts results in the acquisition of a systemic sclerosis�like phenotype (see Glossary) in the production of extracellular matrix proteins (18). Furthermore, antibodies to retroviral proteins have been detected in serum specimens from patients with systemic sclerosis (19).
Another hypothesis has suggested that human cytomegalovirus may be involved in the initial events of systemic sclerosis. This hypothesis is supported by the observations of a higher prevalence of IgA antihuman cytomegalovirus antibodies in patients with systemic sclerosis, which are capable of inducing apoptosis (see Glossary) in human endothelial cells; the increased prevalence of anticytomegalovirus IgA antibodies in patients positive for Scl-70 autoantibodies; and the severe fibroproliferative vascular changes and the increased occurrence of antinuclear antibodies with an immunofluorescence pattern similar to that present in serum specimens from patients with systemic sclerosis in human cytomegalovirus infections (20, 21). Despite intensive study, however, there is no definitive evidence to conclude that systemic sclerosis has a viral origin.
Environmental agents have also been implicated in the development of systemic sclerosis (22, 23). Silica and metal dust exposure had been shown in case studies to be related to systemic sclerosis (22-24), although some studies have failed to confirm an association. On the other hand, organic solvent exposure may eventually be proven to be an important environmental factor in triggering this disease. Indeed, persons exposed to vinyl chloride have an increased risk for skin thickening, the Raynaud phenomenon, and digital ulcers (23), and recent epidemiologic studies have found a higher frequency of organic solvent exposure in patients with systemic sclerosis than in normal controls (24). Several other environmental exposures have been associated with the development of systemic sclerosis, including certain pesticides, hair dyes, and fuel-derived or industrial fumes (22, 23).
The cause of systemic sclerosis has remained elusive despite intense investigations. Although the disease is not inherited in a classical Mendelian pattern, there is strong evidence that genetic factors contribute to its development and clinical manifestations, as discussed in more detail shortly. However, it has become apparent that environmental agents play a crucial and more important role than genetic influences. One study reported a remarkably low concordance in the development of systemic sclerosis among homozygous twins, indicating that the heritability component of the disease was very low and that the most important factor was of an environmental or acquired origin (17). Many infectious, chemical, and physical agents have been postulated as being involved in the cause of the disease. The hypothesis that infectious agents may cause systemic sclerosis has been studied extensively. Some researchers have suggested that the production of specific autoantibodies in systemic sclerosis is the result of an antigen-driven response caused by "molecular mimicry." The concept of "molecular mimicry" proposes that antibodies against self-antigens are produced because these antigens contain epitopes (see Glossary) that share structural similarities with viral or bacterial proteins. In the immunopathogenesis of systemic sclerosis, herpesviruses, retroviruses, and human cytomegalovirus infections, among others, have been suggested as possible causative agents. Evidence supporting the role of retroviruses includes the demonstration of sequence homologies (see Glossary) between certain retroviral proteins and the topoisomerase I antigen (see Glossary), which is the target of anti�Scl-70 antibodies in patients with systemic sclerosis (18). In addition, it has been shown that the induced expression of retroviral proteins in normal human dermal fibroblasts results in the acquisition of a systemic sclerosis�like phenotype (see Glossary) in the production of extracellular matrix proteins (18). Furthermore, antibodies to retroviral proteins have been detected in serum specimens from patients with systemic sclerosis (19).
Another hypothesis has suggested that human cytomegalovirus may be involved in the initial events of systemic sclerosis. This hypothesis is supported by the observations of a higher prevalence of IgA antihuman cytomegalovirus antibodies in patients with systemic sclerosis, which are capable of inducing apoptosis (see Glossary) in human endothelial cells; the increased prevalence of anticytomegalovirus IgA antibodies in patients positive for Scl-70 autoantibodies; and the severe fibroproliferative vascular changes and the increased occurrence of antinuclear antibodies with an immunofluorescence pattern similar to that present in serum specimens from patients with systemic sclerosis in human cytomegalovirus infections (20, 21). Despite intensive study, however, there is no definitive evidence to conclude that systemic sclerosis has a viral origin.
Environmental agents have also been implicated in the development of systemic sclerosis (22, 23). Silica and metal dust exposure had been shown in case studies to be related to systemic sclerosis (22-24), although some studies have failed to confirm an association. On the other hand, organic solvent exposure may eventually be proven to be an important environmental factor in triggering this disease. Indeed, persons exposed to vinyl chloride have an increased risk for skin thickening, the Raynaud phenomenon, and digital ulcers (23), and recent epidemiologic studies have found a higher frequency of organic solvent exposure in patients with systemic sclerosis than in normal controls (24). Several other environmental exposures have been associated with the development of systemic sclerosis, including certain pesticides, hair dyes, and fuel-derived or industrial fumes (22, 23).
18. Systemic sclerosis: pathogenesis Possible Causative Agents
Environmental agents
Silica and metal dust exposure
organic solvent exposure (vinyl chloride)
higher frequency of organic solvent exposure in patients with systemic sclerosis than in normal controls
pesticides, hair dyes, and fuel-derived or industrial fumes The cause of systemic sclerosis has remained elusive despite intense investigations. Although the disease is not inherited in a classical Mendelian pattern, there is strong evidence that genetic factors contribute to its development and clinical manifestations, as discussed in more detail shortly. However, it has become apparent that environmental agents play a crucial and more important role than genetic influences. One study reported a remarkably low concordance in the development of systemic sclerosis among homozygous twins, indicating that the heritability component of the disease was very low and that the most important factor was of an environmental or acquired origin (17). Many infectious, chemical, and physical agents have been postulated as being involved in the cause of the disease. The hypothesis that infectious agents may cause systemic sclerosis has been studied extensively. Some researchers have suggested that the production of specific autoantibodies in systemic sclerosis is the result of an antigen-driven response caused by "molecular mimicry." The concept of "molecular mimicry" proposes that antibodies against self-antigens are produced because these antigens contain epitopes (see Glossary) that share structural similarities with viral or bacterial proteins. In the immunopathogenesis of systemic sclerosis, herpesviruses, retroviruses, and human cytomegalovirus infections, among others, have been suggested as possible causative agents. Evidence supporting the role of retroviruses includes the demonstration of sequence homologies (see Glossary) between certain retroviral proteins and the topoisomerase I antigen (see Glossary), which is the target of anti�Scl-70 antibodies in patients with systemic sclerosis (18). In addition, it has been shown that the induced expression of retroviral proteins in normal human dermal fibroblasts results in the acquisition of a systemic sclerosis�like phenotype (see Glossary) in the production of extracellular matrix proteins (18). Furthermore, antibodies to retroviral proteins have been detected in serum specimens from patients with systemic sclerosis (19).
Another hypothesis has suggested that human cytomegalovirus may be involved in the initial events of systemic sclerosis. This hypothesis is supported by the observations of a higher prevalence of IgA antihuman cytomegalovirus antibodies in patients with systemic sclerosis, which are capable of inducing apoptosis (see Glossary) in human endothelial cells; the increased prevalence of anticytomegalovirus IgA antibodies in patients positive for Scl-70 autoantibodies; and the severe fibroproliferative vascular changes and the increased occurrence of antinuclear antibodies with an immunofluorescence pattern similar to that present in serum specimens from patients with systemic sclerosis in human cytomegalovirus infections (20, 21). Despite intensive study, however, there is no definitive evidence to conclude that systemic sclerosis has a viral origin.
Environmental agents have also been implicated in the development of systemic sclerosis (22, 23). Silica and metal dust exposure had been shown in case studies to be related to systemic sclerosis (22-24), although some studies have failed to confirm an association. On the other hand, organic solvent exposure may eventually be proven to be an important environmental factor in triggering this disease. Indeed, persons exposed to vinyl chloride have an increased risk for skin thickening, the Raynaud phenomenon, and digital ulcers (23), and recent epidemiologic studies have found a higher frequency of organic solvent exposure in patients with systemic sclerosis than in normal controls (24). Several other environmental exposures have been associated with the development of systemic sclerosis, including certain pesticides, hair dyes, and fuel-derived or industrial fumes (22, 23).
The cause of systemic sclerosis has remained elusive despite intense investigations. Although the disease is not inherited in a classical Mendelian pattern, there is strong evidence that genetic factors contribute to its development and clinical manifestations, as discussed in more detail shortly. However, it has become apparent that environmental agents play a crucial and more important role than genetic influences. One study reported a remarkably low concordance in the development of systemic sclerosis among homozygous twins, indicating that the heritability component of the disease was very low and that the most important factor was of an environmental or acquired origin (17). Many infectious, chemical, and physical agents have been postulated as being involved in the cause of the disease. The hypothesis that infectious agents may cause systemic sclerosis has been studied extensively. Some researchers have suggested that the production of specific autoantibodies in systemic sclerosis is the result of an antigen-driven response caused by "molecular mimicry." The concept of "molecular mimicry" proposes that antibodies against self-antigens are produced because these antigens contain epitopes (see Glossary) that share structural similarities with viral or bacterial proteins. In the immunopathogenesis of systemic sclerosis, herpesviruses, retroviruses, and human cytomegalovirus infections, among others, have been suggested as possible causative agents. Evidence supporting the role of retroviruses includes the demonstration of sequence homologies (see Glossary) between certain retroviral proteins and the topoisomerase I antigen (see Glossary), which is the target of anti�Scl-70 antibodies in patients with systemic sclerosis (18). In addition, it has been shown that the induced expression of retroviral proteins in normal human dermal fibroblasts results in the acquisition of a systemic sclerosis�like phenotype (see Glossary) in the production of extracellular matrix proteins (18). Furthermore, antibodies to retroviral proteins have been detected in serum specimens from patients with systemic sclerosis (19).
Another hypothesis has suggested that human cytomegalovirus may be involved in the initial events of systemic sclerosis. This hypothesis is supported by the observations of a higher prevalence of IgA antihuman cytomegalovirus antibodies in patients with systemic sclerosis, which are capable of inducing apoptosis (see Glossary) in human endothelial cells; the increased prevalence of anticytomegalovirus IgA antibodies in patients positive for Scl-70 autoantibodies; and the severe fibroproliferative vascular changes and the increased occurrence of antinuclear antibodies with an immunofluorescence pattern similar to that present in serum specimens from patients with systemic sclerosis in human cytomegalovirus infections (20, 21). Despite intensive study, however, there is no definitive evidence to conclude that systemic sclerosis has a viral origin.
Environmental agents have also been implicated in the development of systemic sclerosis (22, 23). Silica and metal dust exposure had been shown in case studies to be related to systemic sclerosis (22-24), although some studies have failed to confirm an association. On the other hand, organic solvent exposure may eventually be proven to be an important environmental factor in triggering this disease. Indeed, persons exposed to vinyl chloride have an increased risk for skin thickening, the Raynaud phenomenon, and digital ulcers (23), and recent epidemiologic studies have found a higher frequency of organic solvent exposure in patients with systemic sclerosis than in normal controls (24). Several other environmental exposures have been associated with the development of systemic sclerosis, including certain pesticides, hair dyes, and fuel-derived or industrial fumes (22, 23).
19. Systemic sclerosis: pathogenesis Role of Genetic Factors
familial clustering of the disease
high frequency of autoimmune disorders and autoantibodies in family members
differences in prevalence and clinical manifestations among different ethnic groups Role of Genetic Factors
The contribution of genetic factors in the development and expression of systemic sclerosis is strongly supported by the observation of familial clustering of the disease, the high frequency of autoimmune disorders and autoantibodies in family members of patients with systemic sclerosis, differences in prevalence and clinical manifestations among different ethnic groups, and the increased prevalence of certain HLAs and MHC alleles (see Glossary) among different ethnic groups and among patients with different clinical subsets of the disease or with different patterns of autoantibodies (25). Strong evidence indicates that genetic factors largely determine the production of specific autoantibodies in systemic sclerosis. Although the concordance of systemic sclerosis among identical twins is only 4.2% and is not significantly different from the concordance of disease in dizygotic twins (5.9%), the concordance for the presence of specific autoantibodies is substantially higher (17). These observations indicate that inherited genetic factors are important for the production of autoantibodies but are not sufficient for development of disease.
Prevalence of the disease varies by geographic region and by ethnic background; the prevalence in the United States is 242 to 286 cases per million persons in the population. However, in Native American persons of the Choctaw tribe in Oklahoma, one of the best-studied groups for the role of genetic factors in systemic sclerosis, the prevalence is 469 cases per million persons in the population (26). The differences of MHC and HLA allele expression are evident when comparing the different haplotypes (see Glossary) identified as being linked to disease expression, in particular to the pattern of autoantibody response, among ethnic groups (27). Disease expression also appears to differ among ethnic groups. African-American persons are more likely to have anti-topoisomerase I antibodies and more severe visceral manifestations, including a higher frequency of pulmonary fibrosis. In contrast, anticentromere antibodies are more common in white persons, who are also more likely to have limited disease with less severe systemic manifestations (27).Role of Genetic Factors
The contribution of genetic factors in the development and expression of systemic sclerosis is strongly supported by the observation of familial clustering of the disease, the high frequency of autoimmune disorders and autoantibodies in family members of patients with systemic sclerosis, differences in prevalence and clinical manifestations among different ethnic groups, and the increased prevalence of certain HLAs and MHC alleles (see Glossary) among different ethnic groups and among patients with different clinical subsets of the disease or with different patterns of autoantibodies (25). Strong evidence indicates that genetic factors largely determine the production of specific autoantibodies in systemic sclerosis. Although the concordance of systemic sclerosis among identical twins is only 4.2% and is not significantly different from the concordance of disease in dizygotic twins (5.9%), the concordance for the presence of specific autoantibodies is substantially higher (17). These observations indicate that inherited genetic factors are important for the production of autoantibodies but are not sufficient for development of disease.
Prevalence of the disease varies by geographic region and by ethnic background; the prevalence in the United States is 242 to 286 cases per million persons in the population. However, in Native American persons of the Choctaw tribe in Oklahoma, one of the best-studied groups for the role of genetic factors in systemic sclerosis, the prevalence is 469 cases per million persons in the population (26). The differences of MHC and HLA allele expression are evident when comparing the different haplotypes (see Glossary) identified as being linked to disease expression, in particular to the pattern of autoantibody response, among ethnic groups (27). Disease expression also appears to differ among ethnic groups. African-American persons are more likely to have anti-topoisomerase I antibodies and more severe visceral manifestations, including a higher frequency of pulmonary fibrosis. In contrast, anticentromere antibodies are more common in white persons, who are also more likely to have limited disease with less severe systemic manifestations (27).
20. Systemic sclerosis: pathogenesis increased prevalence of certain HLAs and MHC alleles among different ethnic groups and among patients with different clinical subsets of the disease or with different patterns of autoantibodies
Choctaw tribe, Oklahoma (double the prevalence)
African-Americans- greater likelihood anti-topoisomerase I abs and more severe visceral manifestations
anticentromere antibodies are more common in caucasians Role of Genetic Factors
The contribution of genetic factors in the development and expression of systemic sclerosis is strongly supported by the observation of familial clustering of the disease, the high frequency of autoimmune disorders and autoantibodies in family members of patients with systemic sclerosis, differences in prevalence and clinical manifestations among different ethnic groups, and the increased prevalence of certain HLAs and MHC alleles (see Glossary) among different ethnic groups and among patients with different clinical subsets of the disease or with different patterns of autoantibodies (25). Strong evidence indicates that genetic factors largely determine the production of specific autoantibodies in systemic sclerosis. Although the concordance of systemic sclerosis among identical twins is only 4.2% and is not significantly different from the concordance of disease in dizygotic twins (5.9%), the concordance for the presence of specific autoantibodies is substantially higher (17). These observations indicate that inherited genetic factors are important for the production of autoantibodies but are not sufficient for development of disease.
Prevalence of the disease varies by geographic region and by ethnic background; the prevalence in the United States is 242 to 286 cases per million persons in the population. However, in Native American persons of the Choctaw tribe in Oklahoma, one of the best-studied groups for the role of genetic factors in systemic sclerosis, the prevalence is 469 cases per million persons in the population (26). The differences of MHC and HLA allele expression are evident when comparing the different haplotypes (see Glossary) identified as being linked to disease expression, in particular to the pattern of autoantibody response, among ethnic groups (27). Disease expression also appears to differ among ethnic groups. African-American persons are more likely to have anti-topoisomerase I antibodies and more severe visceral manifestations, including a higher frequency of pulmonary fibrosis. In contrast, anticentromere antibodies are more common in white persons, who are also more likely to have limited disease with less severe systemic manifestations (27).Role of Genetic Factors
The contribution of genetic factors in the development and expression of systemic sclerosis is strongly supported by the observation of familial clustering of the disease, the high frequency of autoimmune disorders and autoantibodies in family members of patients with systemic sclerosis, differences in prevalence and clinical manifestations among different ethnic groups, and the increased prevalence of certain HLAs and MHC alleles (see Glossary) among different ethnic groups and among patients with different clinical subsets of the disease or with different patterns of autoantibodies (25). Strong evidence indicates that genetic factors largely determine the production of specific autoantibodies in systemic sclerosis. Although the concordance of systemic sclerosis among identical twins is only 4.2% and is not significantly different from the concordance of disease in dizygotic twins (5.9%), the concordance for the presence of specific autoantibodies is substantially higher (17). These observations indicate that inherited genetic factors are important for the production of autoantibodies but are not sufficient for development of disease.
Prevalence of the disease varies by geographic region and by ethnic background; the prevalence in the United States is 242 to 286 cases per million persons in the population. However, in Native American persons of the Choctaw tribe in Oklahoma, one of the best-studied groups for the role of genetic factors in systemic sclerosis, the prevalence is 469 cases per million persons in the population (26). The differences of MHC and HLA allele expression are evident when comparing the different haplotypes (see Glossary) identified as being linked to disease expression, in particular to the pattern of autoantibody response, among ethnic groups (27). Disease expression also appears to differ among ethnic groups. African-American persons are more likely to have anti-topoisomerase I antibodies and more severe visceral manifestations, including a higher frequency of pulmonary fibrosis. In contrast, anticentromere antibodies are more common in white persons, who are also more likely to have limited disease with less severe systemic manifestations (27).
21. Systemic sclerosis: pathogenesis Microchimerism
during pregnancy, allogenic fetal or maternal cells cross the placenta in bidirectional traffic and persist in the circulation and tissues of the mother or child
These engrafted foreign cells may become activated by a second event and may mount a graft-versus-host reaction, which manifests as systemic sclerosis
similarities in clinical, histopathologic, and serologic features between graft-versus-host disease and systemic sclerosis Microchimerism is a novel and provocative hypothesis of the cause of systemic sclerosis (32-34). The hypothesis suggests that, during pregnancy, allogenic (see Glossary) fetal or maternal cells cross the placenta in bidirectional traffic and persist in the circulation and tissues of the mother or child, respectively, as a result of HLA II (DRB1) compatibility between the mother and the fetus. These engrafted foreign cells may become activated by a second event and may mount a graft-versus-host reaction, which manifests as systemic sclerosis. The remarkable similarities in clinical, histopathologic, and serologic features between graft-versus-host disease and systemic sclerosis, including esophageal, lung, and skin involvement; lymphocytic infiltration and fibrosis of affected organs; and production of autoantibodies, strongly support this hypothesis. Microchimerism is a novel and provocative hypothesis of the cause of systemic sclerosis (32-34). The hypothesis suggests that, during pregnancy, allogenic (see Glossary) fetal or maternal cells cross the placenta in bidirectional traffic and persist in the circulation and tissues of the mother or child, respectively, as a result of HLA II (DRB1) compatibility between the mother and the fetus. These engrafted foreign cells may become activated by a second event and may mount a graft-versus-host reaction, which manifests as systemic sclerosis. The remarkable similarities in clinical, histopathologic, and serologic features between graft-versus-host disease and systemic sclerosis, including esophageal, lung, and skin involvement; lymphocytic infiltration and fibrosis of affected organs; and production of autoantibodies, strongly support this hypothesis.
22. Systemic sclerosis: pathogenesis Humoral Immune System Alterations
>90% of patients with SSc Have + ANA
Anti�Scl-70 abs (antibodies against DNA topoisomerase I) in 30- 40% of pts with diffuse SSc
Anticentromere abs in 80% to 90% of patients with limited SSc; < 10% of patients with diffuse SSc
Humoral Immune System Alterations
The presence of specific autoantibodies is one of the most common manifestations of systemic sclerosis. More than 90% of patients with systemic sclerosis harbor antinuclear antibodies in their serum. Numerous autoantibodies, some of which are extremely specific for systemic sclerosis, have been described in patients with the disease; other autoantibodies are associated with different clinical manifestations (38).
Anti�Scl-70 antibodies have been shown to react with DNA topoisomerase I and are almost exclusively present in serum specimens from patients with the diffuse form of systemic sclerosis, although only about 30% to 40% of these patients harbor these autoantibodies. Anticentromere antibodies are present in 80% to 90% of patients with the limited form of systemic sclerosis but are found in fewer than 10% of patients with diffuse systemic sclerosis. These 2 autoantibodies rarely coexist in the same patient.
Other autoantibodies are less common in patients with systemic sclerosis. They include anti-RNA polymerases I and III antibodies in patients with rapidly progressive disease and severe internal organ involvement; antifibrillarin antibodies, which are commonly found in diffuse systemic sclerosis; and anti�PM-Scl antibodies, which are usually found in patients with systemic sclerosis who develop an inflammatory myopathy. Although autoantibodies are common in systemic sclerosis, they are not directly involved in the clinical manifestations of the disease. However, owing to their high frequency and their specificity for certain clinical disease subsets, their presence is very helpful in establishing the diagnosis and predicting a probable pattern of organ involvement, severity, and disease progression (38).
Humoral Immune System Alterations
The presence of specific autoantibodies is one of the most common manifestations of systemic sclerosis. More than 90% of patients with systemic sclerosis harbor antinuclear antibodies in their serum. Numerous autoantibodies, some of which are extremely specific for systemic sclerosis, have been described in patients with the disease; other autoantibodies are associated with different clinical manifestations (38).
Anti�Scl-70 antibodies have been shown to react with DNA topoisomerase I and are almost exclusively present in serum specimens from patients with the diffuse form of systemic sclerosis, although only about 30% to 40% of these patients harbor these autoantibodies. Anticentromere antibodies are present in 80% to 90% of patients with the limited form of systemic sclerosis but are found in fewer than 10% of patients with diffuse systemic sclerosis. These 2 autoantibodies rarely coexist in the same patient.
Other autoantibodies are less common in patients with systemic sclerosis. They include anti-RNA polymerases I and III antibodies in patients with rapidly progressive disease and severe internal organ involvement; antifibrillarin antibodies, which are commonly found in diffuse systemic sclerosis; and anti�PM-Scl antibodies, which are usually found in patients with systemic sclerosis who develop an inflammatory myopathy. Although autoantibodies are common in systemic sclerosis, they are not directly involved in the clinical manifestations of the disease. However, owing to their high frequency and their specificity for certain clinical disease subsets, their presence is very helpful in establishing the diagnosis and predicting a probable pattern of organ involvement, severity, and disease progression (38).
23. Scleroderma Sclero �hard� Derma �skin�
Chronic multisystem disease
20/million
4-5x�s more common in females
Avg age 50yo
Caucasians milder limited disease
African Americans more severe disease
24. Differential Diagnosis of SSc Skin thickening
Bleomycin fibrosis
Digital CRPS
Mycosis fungoids
Amyloidosis
Vinyl chloride disease
Celiac disease
Acrodermatitis chronica atrophicans
Skin thickening sparing fingers/hands
Scleromyedema
Eosinophilic fascitis
Eosinophilic myalgia syndrome
Graft vs host
Amyloidosis
Porphyria cutanea tarda
25. Systemic sclerosis Clinical features:
Chronic multisystem disease
Initial symptoms: raynaud�s, fatigue, musculoskeletal complaints, �puffiness� of the hands
Significant skin, cardiac, pulmonary, GI, renal disease can subsequently develop
Classification
Limited SSc
Diffuse SSc
26. Systemic sclerosis Classification
Limited SSc
Skin thickening limited to symmetrical changes of fingers, distal arms, face/neck
Progression of disease after onset of Raynaud�s
CREST
Associated with anticentromere Ab
Relatively good prognosis with survival >70% 10yrs
27. Systemic sclerosis Classification
Diffuse SSc
Proximal skin thickening
Rapid onset of disease following appearance of Raynaud�s
Significant visceral disease (lung, CV, GI, renal)
Associated with ANA, no anticentromere Ab
Variable disease course but overall poor prognosis
Survival 40-60% at 10 yrs
28. Systemic sclerosis Classification
Undefined connective tissue disease
Patients with features of systemic sclerosis, but who do not have definite clinical/ laboratory findings
Localized scleroderma
Morphea: plaques of fibrotic skin and subcutaneous tissue without systemic disease
Linear scleroderma: longitudinal fibrotic bands that occur predominantly on extremities and involve skin and deeper tissues
29. Clinical Features Raynaud�s Phenomenon:
Pts complain of cold hands/ feet, associated with triphasic color changes of the digits
Can occur up to 10 yrs prior to the development of other features of SSc
90% of pts with SSc have raynaud�s
If raynaud�s occurs after age 20, there is greater likelihood that it is secondary to other underlying disorder
31. Clinical Features Skin changes:
Early: skin appears inflamed with non- pitting edema, erythema, pruritus, followed by increased collagen deposition, hypo/ hyper- pigmentary changes
Fibrotic stage: skin more thickened, pruritic
Atrophic stage: skin atrophic, thinned, ulcerations develop
Other: dilated capillary loops, loss of capillaries in the skin of the nail folds, telangiectasias, calcium hydroxyapatite deposition
37. Clinical features Musculoskeletal
Early: non-specific arthralgias, myalgias, out of proportion to physical exam findings
Friction rubs (in diffuse skin disease)
38. Clinical features Pulmonary
Leading cause of mortality
Interstitial lung disease
Can manifest as dyspnea on exertion or can be asymptomatic found on routine imaging
Fine crackles on physical exam
PFT�s and Ct chest may show early changes
Restrictive pattern on PFT
Pulmonary vascular disease (PAH)
Pulmonary vascular disease w or w/o fibrosis
More common limited
40. Clinical Features Gastrointestinal
In diffuse and limited SSc
Small oral aperture, dry MM, periodontal disease
Dysphasia, GERD, esophagitis
Early esophageal dysmotility is likely due to neuromuscular dysfunction, later due to collagen deposition, smooth mm atrophy, fibrosis
41. Clinical Features Gastrointestinal
�Watermelon stomach�
a condition in which the lining of the stomach bleeds, causing it to look like the characteristic stripes of a watermelon when viewed by Endoscopy.
Constipation
Pseudo-obstruction
Gastroparesis
43. Clinical Features Gastrointestinal (cont)
Poor gastric emptying
Dysmotility of small intestine
Large bowel: wide mouth diverticuli, fecal incontinence from fibrosis of rectal sphincters
46. Clinical Features Cardiac
Usually not appreciated until late in the disease
DOE, palpitations, chest pain
Patchy fibrosis throughout myocardium
Vascular heart disease
Diastolic dysfunction is frequent
Cardiomyopathy as a result of associated autoimmune myocarditis
Pericardial effusions (30- 40%)
Conduction system disease
47. Clinical Features Renal
SSc renal crisis: accelerated HTN, rapidly progressive renal failure
80% of cases of renal cell crisis occur within first 4-5 years of disease (usually diffuse disease)
Findings: HTN, elevated Cr, proteinuria, microscopic hematuria, anemia, thrombocytopenia,
Treatment: ACE inhibitors
49. Clinical Features Other signs:
Depression
Sexual dysfunction
Associated sjogren�s
CTS
Thyroid disease (thyroid fibrosis or autoimmune thyroiditis)
Primary biliary cirrhosis, autoimmune hepatitis associated with CREST syndrome (rare)
50. Localized scleroderma:
Circumscribed fibrotic areas involving the skin
More common in females, children
Clinical course is benign- no internal organ involvement
Non-specific area erythema, pain, with ivory fibrotic center, and surrounding margin of hypopigmentation
�en coup de sabre�
Types: isolated morphea, generalized morphea, linear scleroderma, nodular scleroderma, localized bullous lesions
54. Systemic sclerosis: treatment Treatment of vascular damage
Prevention of fibrosis
Suppression of immune response
Rational treatment of systemic sclerosis
The rational treatment of systemic sclerosis should be based on a hypothesis of its pathogenesis. Such a hypothesis was proposed by Furst (Fig. 1)�[Not Available] [11] . Although the etiology of SSc remains to be elucidated, most experts believe that it is probably multifactorial. Genetic susceptibility predisposes an individual to some environmental stimuli. This can be seen in Choctaw Indians in Oklahoma, who are known to have a high prevalence of systemic sclerosis. The combination of genetic and environmental factors may lead to activation of the immune system or result in endothelial injury.
Multiple inflammatory mediators are probably directly involved in the pathogenesis of systemic sclerosis which results in activation and proliferation of fibroblasts. Cytokines that are released by the activated immune systems include transforming growth factor-beta (TGF-�), connective tissue growth factor (CTGF), and platelet-derived growth factor. If the endothelium is affected or damaged, then other cytokines, such as intracellular adhesion molecule (ICAM), endolethial lymphocyte adhesion molecule (ELAM), and vascular endolethic growth factor (VEGF) and endothelin-1 are released, which result in immune activation and fibroblast proliferation. The proliferation of fibroblasts eventually leads to collagen accumulation and end-organ damage, which defines the phenotypic expression of systemic sclerosis.
Because such a pathogenetic construct is reasonable, one can develop a rational approach to treat systemic sclerosis by interrupting the pathogenetic cycle. Treatment might, for example, be aimed at the vascular damage, prevention of fibrosis, or suppression of the immune response.
Rational treatment of systemic sclerosis
The rational treatment of systemic sclerosis should be based on a hypothesis of its pathogenesis. Such a hypothesis was proposed by Furst (Fig. 1)�[Not Available] [11] . Although the etiology of SSc remains to be elucidated, most experts believe that it is probably multifactorial. Genetic susceptibility predisposes an individual to some environmental stimuli. This can be seen in Choctaw Indians in Oklahoma, who are known to have a high prevalence of systemic sclerosis. The combination of genetic and environmental factors may lead to activation of the immune system or result in endothelial injury.
Multiple inflammatory mediators are probably directly involved in the pathogenesis of systemic sclerosis which results in activation and proliferation of fibroblasts. Cytokines that are released by the activated immune systems include transforming growth factor-beta (TGF-�), connective tissue growth factor (CTGF), and platelet-derived growth factor. If the endothelium is affected or damaged, then other cytokines, such as intracellular adhesion molecule (ICAM), endolethial lymphocyte adhesion molecule (ELAM), and vascular endolethic growth factor (VEGF) and endothelin-1 are released, which result in immune activation and fibroblast proliferation. The proliferation of fibroblasts eventually leads to collagen accumulation and end-organ damage, which defines the phenotypic expression of systemic sclerosis.
Because such a pathogenetic construct is reasonable, one can develop a rational approach to treat systemic sclerosis by interrupting the pathogenetic cycle. Treatment might, for example, be aimed at the vascular damage, prevention of fibrosis, or suppression of the immune response.
55. Systemic sclerosis: treatment Kidneys:
ACE inhibitors for SSc renal crisis
Lungs
Pulmonary fibrosis:
Cyclophosphamide
Summary
Treatment of systemic sclerosis has been somewhat haphazard and treatment has often been �borrowed� from the experience gained from treating other connective tissue diseases. There was a period of time that was focused mainly on organ-specific manifestations of systemic sclerosis and some advance in preventing vital organ damage (such as renal crisis) was achieved. The vast improvement in mortality from the use of ACE inhibitors raises one's hopes for other effective therapeutic interventions.
At this juncture, the evidence is strong that the ACE inhibitors that are used in scleroderma renal crisis are disease-modifying, even without proving it by a randomized controlled trial. The evidence is strong that the use of epoprostenol for primary pulmonary hypertension is life-saving; however, whether epoprostenol is life-saving in the pulmonary hypertension in scleroderma remains to be proven. There are suggestions that bosentan (for the pulmonary hypertension of scleroderma), cyclophosphamide (for SSc alveolitis), stem cell transplant, interferon-? (for interstitial pulmonary fibrosis), and methotrexate (for the skin thickening of diffuse scleroderma) may improve organ function or functional activities, but whether they are truly disease-modifying remains to be proven.
As we increase our understanding of the pathophysiology of systemic sclerosis and we learn how better to design trials for systemic sclerosis, we may be more successful in developing optimal disease-modifying therapy. Although the treatment of systemic sclerosis remains difficult, there are an increasing number of potentially effective regimens that are undergoing clinical investigations. A rational approach to therapy seems possible, based on a hypothesis of the pathogenesis of systemic sclerosis. Thus, there is accumulating evidence that supports the use of prostacyclin derivatives to treat systemic sclerosis, some evidence that antifibrotic regimens may be effective, and moderate evidence that immunosuppression also may be effective in certain stages of this disease.
Summary
Treatment of systemic sclerosis has been somewhat haphazard and treatment has often been �borrowed� from the experience gained from treating other connective tissue diseases. There was a period of time that was focused mainly on organ-specific manifestations of systemic sclerosis and some advance in preventing vital organ damage (such as renal crisis) was achieved. The vast improvement in mortality from the use of ACE inhibitors raises one's hopes for other effective therapeutic interventions.
At this juncture, the evidence is strong that the ACE inhibitors that are used in scleroderma renal crisis are disease-modifying, even without proving it by a randomized controlled trial. The evidence is strong that the use of epoprostenol for primary pulmonary hypertension is life-saving; however, whether epoprostenol is life-saving in the pulmonary hypertension in scleroderma remains to be proven. There are suggestions that bosentan (for the pulmonary hypertension of scleroderma), cyclophosphamide (for SSc alveolitis), stem cell transplant, interferon-? (for interstitial pulmonary fibrosis), and methotrexate (for the skin thickening of diffuse scleroderma) may improve organ function or functional activities, but whether they are truly disease-modifying remains to be proven.
As we increase our understanding of the pathophysiology of systemic sclerosis and we learn how better to design trials for systemic sclerosis, we may be more successful in developing optimal disease-modifying therapy. Although the treatment of systemic sclerosis remains difficult, there are an increasing number of potentially effective regimens that are undergoing clinical investigations. A rational approach to therapy seems possible, based on a hypothesis of the pathogenesis of systemic sclerosis. Thus, there is accumulating evidence that supports the use of prostacyclin derivatives to treat systemic sclerosis, some evidence that antifibrotic regimens may be effective, and moderate evidence that immunosuppression also may be effective in certain stages of this disease.
56. Systemic sclerosis: treatment PAH
Epoprostenol (Flolan)
Bosentan (tracleer)- endothelin antagonist
Sildanafil (Viagra/Revatio) Inhibits phosphodiesterase type 5 (PDE-5) in smooth muscle of pulmonary vasculature causing vasodilation Summary
Treatment of systemic sclerosis has been somewhat haphazard and treatment has often been �borrowed� from the experience gained from treating other connective tissue diseases. There was a period of time that was focused mainly on organ-specific manifestations of systemic sclerosis and some advance in preventing vital organ damage (such as renal crisis) was achieved. The vast improvement in mortality from the use of ACE inhibitors raises one's hopes for other effective therapeutic interventions.
At this juncture, the evidence is strong that the ACE inhibitors that are used in scleroderma renal crisis are disease-modifying, even without proving it by a randomized controlled trial. The evidence is strong that the use of epoprostenol for primary pulmonary hypertension is life-saving; however, whether epoprostenol is life-saving in the pulmonary hypertension in scleroderma remains to be proven. There are suggestions that bosentan (for the pulmonary hypertension of scleroderma), cyclophosphamide (for SSc alveolitis), stem cell transplant, interferon-? (for interstitial pulmonary fibrosis), and methotrexate (for the skin thickening of diffuse scleroderma) may improve organ function or functional activities, but whether they are truly disease-modifying remains to be proven.
As we increase our understanding of the pathophysiology of systemic sclerosis and we learn how better to design trials for systemic sclerosis, we may be more successful in developing optimal disease-modifying therapy. Although the treatment of systemic sclerosis remains difficult, there are an increasing number of potentially effective regimens that are undergoing clinical investigations. A rational approach to therapy seems possible, based on a hypothesis of the pathogenesis of systemic sclerosis. Thus, there is accumulating evidence that supports the use of prostacyclin derivatives to treat systemic sclerosis, some evidence that antifibrotic regimens may be effective, and moderate evidence that immunosuppression also may be effective in certain stages of this disease.
Summary
Treatment of systemic sclerosis has been somewhat haphazard and treatment has often been �borrowed� from the experience gained from treating other connective tissue diseases. There was a period of time that was focused mainly on organ-specific manifestations of systemic sclerosis and some advance in preventing vital organ damage (such as renal crisis) was achieved. The vast improvement in mortality from the use of ACE inhibitors raises one's hopes for other effective therapeutic interventions.
At this juncture, the evidence is strong that the ACE inhibitors that are used in scleroderma renal crisis are disease-modifying, even without proving it by a randomized controlled trial. The evidence is strong that the use of epoprostenol for primary pulmonary hypertension is life-saving; however, whether epoprostenol is life-saving in the pulmonary hypertension in scleroderma remains to be proven. There are suggestions that bosentan (for the pulmonary hypertension of scleroderma), cyclophosphamide (for SSc alveolitis), stem cell transplant, interferon-? (for interstitial pulmonary fibrosis), and methotrexate (for the skin thickening of diffuse scleroderma) may improve organ function or functional activities, but whether they are truly disease-modifying remains to be proven.
As we increase our understanding of the pathophysiology of systemic sclerosis and we learn how better to design trials for systemic sclerosis, we may be more successful in developing optimal disease-modifying therapy. Although the treatment of systemic sclerosis remains difficult, there are an increasing number of potentially effective regimens that are undergoing clinical investigations. A rational approach to therapy seems possible, based on a hypothesis of the pathogenesis of systemic sclerosis. Thus, there is accumulating evidence that supports the use of prostacyclin derivatives to treat systemic sclerosis, some evidence that antifibrotic regimens may be effective, and moderate evidence that immunosuppression also may be effective in certain stages of this disease.