Download
1 / 60
600 likes | 606 Vues
Выравнивание двух последовательностей. -4. -6. -2. -3. -4. -2. -6. -3. -2. -8. -5. -4. 0. -2. 1. -1. -1. 0. -1. -1. Sequence comparison: Motivation. Finding similarity between sequences is important for many biological questions. Find homologous proteins

E N D
Выравнивание двух последовательностей
-4 -6 -2 -3 -4 -2 -6 -3 -2 -8 -5 -4 0 -2 1 -1 -1 0 -1 -1
Sequence comparison: Motivation Finding similarity between sequences is important for many biological questions. • Find homologous proteins • Allows to predict structure and function • Locate similar subsequences in DNA • e.g: allows to identify regulatory elements • Locate DNA sequences that might overlap • Helps in sequence assembly
Dot plots Not technically an “alignment” But gives picture of correspondence between pairs of sequences Dot represents similarity between segments of the two sequences
Sequence Alignment • Input: two sequences over the same alphabet • Output: an alignment of the two sequences • Two basic variants of sequence alignment: • Global – all characters in both sequences participate • Needleman-Wunsch, 1970 • Local – find related regions within sequences • Smith-Waterman, 1981
Sequence Alignment - Example -GCGC-ATGGATTGAGCGA TGCGCCATTGAT-GACC-A • Three elements: • Perfect matches • Mismatches • Insertions & deletions (indel) • Input: GCGCATGGATTGAGCGAandTGCGCCATTGATGACCA • Possible output:
Scoring Function • Score each position independently: • Match: +1 • Mismatch: -1 • Indel: -2 • Score of an alignment is sum of position scores • Example: -GCGC-ATGGATTGAGCGA TGCGCCATTGAT-GACC-A Score: (+1x13) + (-1x2) + (-2x4) = 3 ------GCGCATGGATTGAGCGA TGCGCC----ATTGATGACCA-- Score: (+1x5) + (-1x6) + (-2x11) = -23
Sequence vs. Structure Similarity Sequence 1 lcl|1A6M:_ MYOGLOBIN Length 151 (1..151) Sequence 2 lcl|1JL7:A MONOMER HEMOGLOBIN COMPONENT III Length 147 (1..147) Score = 31.6 bits (70), Expect = 10 Identities = 33/137 (24%), Positives = 55/137 (40%), Gaps = 17/137 (12%) Query: 2 LSEGEWQLVLHVWAKVEA--DVAGHGQDILIRLFKSHPETLEKFDRFKHLKTEAEMKASE 59 LS + Q+V W + + AG G++ L + +HPE F + Sbjct: 2 LSAAQRQVVASTWKDIAGADNGAGVGKECLSKFISAHPEMAAVFG--------FSGASDP 53 Query: 60 DLKKHGVTVLTALGAI---LKKKGHHEAELKPLAQSH---ATKHKIPIKYLEFISEAIIH 113 + + G VL +G L +G AE+K + H KH I +Y E + +++ Sbjct: 54 GVAELGAKVLAQIGVAVSHLGDEGKMVAEMKAVGVRHKGYGNKH-IKAEYFEPLGASLLS 112 Query: 114 VLHSRHPGDFGADAQGA 130 + R G A A+ A Sbjct: 113 AMEHRIGGKMNAAAKDA 129
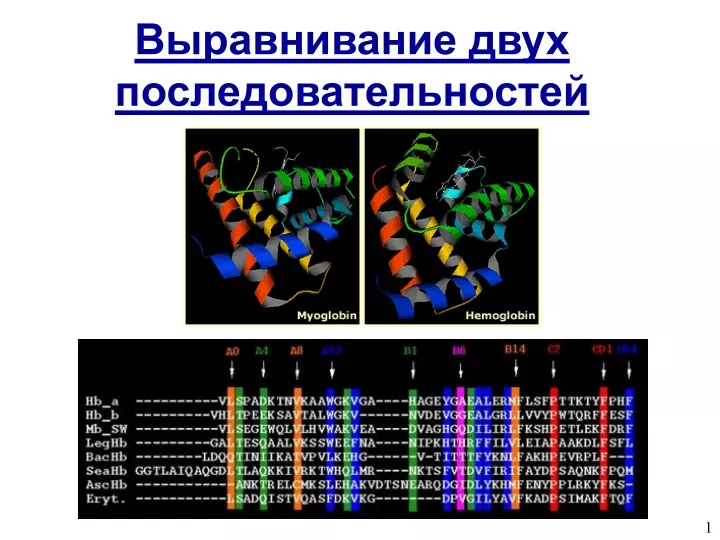
Example Alignment: Globins figure at right shows prototypical structure of globins figure below shows part of alignment for 8 globins (-’s indicate gaps)
Insertions/Deletions and Protein Structure Why is it that two “similar” sequences may have large insertions/deletions? some insertions and deletions may not significantly affect the structure of a protein loop structures: insertions/deletions here not so significant
Sequence vs. Structure Similarity Myoglobin and hemoglobin are similar, but slight differences in structure let them perform different functions. 1A6M: Myoglobin 1JL7: Hemoglobin
Myoglobin & Hemoglobin Красивые ролики по структуре миоглобина и гемоглобина http://higheredbcs.wiley.com/legacy/college/boyer/0471661791/structure/HbMb/hbmb.htm
The Space of Global Alignments some possible global alignments for ELV and VIS ELV VIS -ELV VIS- --ELV VIS-- ELV- -VIS E-LV VIS- ELV-- --VIS EL-V -VIS
Number of Possible Alignments given sequences of length m and n assume we don’t count as distinct and we can have as few as 0 and as many as min{m, n} aligned pairs therefore the number of possible alignments is given by -C G- C- -G
Number of Possible Alignments there are • possible global alignments for 2 sequences of length n • e.g. two sequences of length 100 have possible alignments • but we can use dynamic programming to find an optimal alignment efficiently
Dynamic Programming Algorithmic technique for optimization problems that have two properties: • Optimal substructure: Optimal solution can be computed from optimal solutions to subproblems • Overlapping subproblems: Subproblems overlap such that the total number of distinct subproblems to be solved is relatively small
Dynamic Programming Break problem into overlapping subproblems use memoization: remember solutions to subproblems that we have already seen 3 7 5 1 8 6 2 4
Fibonacci example 1,1,2,3,5,8,13,21,... fib(n) = fib(n - 2) + fib(n - 1) Could implement as a simple recursive function However, complexity of simple recursive function is exponential in n
Fibonacci dynamic programming Two approaches Memoization: Store results from previous calls of function in a table (top down approach) Solve subproblems from smallest to largest, storing results in table (bottom up approach) Both require evaluating all (n-1) subproblems only once: O(n)
Dynamic Programming Graphs 1 2 3 4 5 6 graph for fib(6) Dynamic programming algorithms can be represented by a directed acyclic graph • Each subproblem is a vertex • Direct dependencies between subproblems are edges
Global Alignment • Input: two sequences over the same alphabet • Output: an alignment of the two sequences in which all characters in both sequences participate • The Needleman-Wunsch algorithm finds an optimal global alignment between two sequences • Uses a scoring function • A dynamic programming algorithm
Dynamic Programming Idea consider last step in computing alignment of AAAC with AGC three possible options; in each we’ll choose a different pairing for end of alignment, and add this to the best alignment of previous characters AAA AAA C C AG AGC - C AAAC - AG C consider best alignment of these prefixes score of aligning this pair +
The Needleman-Wunsch (NW) Algorithm • Suppose we have two sequences: • s=s1…sn and t=t1…tm • Construct a matrix V[n+1, m+1] in which V(i, j) contains the score for the best alignment between s1…si and t1…tj. • The grade for cell V(i, j) is: V(i-1, j)+d V(i, j) = max V(i, j-1)+d V(i-1, j-1)+score(si, tj) • d- штраф за открытие разрыва (gap-open) - linear gap penalty • V(n,m) is the score for the best alignment between s and t
NW Algorithm – An Example • Alphabet: • DNA, ∑ = {A,C,G,T} • Input: • s = AAAC • t = AGC • Scoring scheme: • Match: score(x, x) = 1 • Mismatch: score(x, y) = -1 • Gap Opening d = -2 V(i-1, j)+d V(i, j) = max V(i, j-1)+d V(i-1, j-1)+score(si, tj)
Initializing Matrix: Global Alignment with Linear Gap Penalty A G C s 2s 3s 0 A s A 2s A 3s C 4s
-4 -6 -2 -3 -4 -2 -6 -3 -2 -8 -5 -4 NW Algorithm – An Example Match: score(x, x) = 1 Mismatch: score(x, y) = -1 Gap Opening d = -2 V(i-1, j)+d V(i, j) = max V(i, j-1)+d V(i-1, j-1)+score(si, tj) -AGC AAAC 0 -2 1 -1 AG-C AAAC -1 0 -1 A-GC AAAC -1 Лучший вес по определению Обратный проход:движемся обратно по тем ячейкам, из которых было вычислено
-4 -6 -2 -3 -4 -2 -6 -3 -2 -8 -5 -4 NW – Time and Space Complexity Time: • Filling the matrix: • Backtracing: • Overall: Space: • Holding the matrix: 0 -2 O(n·m) 1 -1 O(n+m) O(n·m) -1 0 -1 -1 O(n·m)
Local Alignment Motivation useful for comparing protein sequences that share a common motif (conserved pattern) or domain (independently folded unit) but differ elsewhere useful for comparing DNA sequences that share a similar motif but differ elsewhere useful for comparing protein sequences against genomic DNA sequences (long stretches of uncharacterized sequence) more sensitive when comparing highly diverged sequences
Structure of a genome a gene transcription pre-mRNA splicing mature mRNA translation Human 3x109 bp Genome: ~30,000 genes ~200,000 exons ~23 Mb coding ~15 Mb noncoding protein
Structure of a genome gene D A B C Make D If B then NOT D If A and B then D short sequences regulate expression of genes lots of “junk” sequence e.g. ~50% repeats selfish DNA gene B D C Make B If D then B
Cross-species genome similarity • 98% of genes are conserved between any two mammals • ~75% average similarity in protein sequence hum_a : GTTGACAATAGAGGGTCTGGCAGAGGCTC--------------------- @ 57331/400001 mus_a : GCTGACAATAGAGGGGCTGGCAGAGGCTC--------------------- @ 78560/400001 rat_a : GCTGACAATAGAGGGGCTGGCAGAGACTC--------------------- @ 112658/369938 fug_a : TTTGTTGATGGGGAGCGTGCATTAATTTCAGGCTATTGTTAACAGGCTCG @ 36008/68174 hum_a : CTGGCCGCGGTGCGGAGCGTCTGGAGCGGAGCACGCGCTGTCAGCTGGTG @ 57381/400001 mus_a : CTGGCCCCGGTGCGGAGCGTCTGGAGCGGAGCACGCGCTGTCAGCTGGTG @ 78610/400001 rat_a : CTGGCCCCGGTGCGGAGCGTCTGGAGCGGAGCACGCGCTGTCAGCTGGTG @ 112708/369938 fug_a : TGGGCCGAGGTGTTGGATGGCCTGAGTGAAGCACGCGCTGTCAGCTGGCG @ 36058/68174 hum_a : AGCGCACTCTCCTTTCAGGCAGCTCCCCGGGGAGCTGTGCGGCCACATTT @ 57431/400001 mus_a : AGCGCACTCG-CTTTCAGGCCGCTCCCCGGGGAGCTGAGCGGCCACATTT @ 78659/400001 rat_a : AGCGCACTCG-CTTTCAGGCCGCTCCCCGGGGAGCTGCGCGGCCACATTT @ 112757/369938 fug_a : AGCGCTCGCG------------------------AGTCCCTGCCGTGTCC @ 36084/68174 hum_a : AACACCATCATCACCCCTCCCCGGCCTCCTCAACCTCGGCCTCCTCCTCG @ 57481/400001 mus_a : AACACCGTCGTCA-CCCTCCCCGGCCTCCTCAACCTCGGCCTCCTCCTCG @ 78708/400001 rat_a : AACACCGTCGTCA-CCCTCCCCGGCCTCCTCAACCTCGGCCTCCTCCTCG @ 112806/369938 fug_a : CCGAGGACCCTGA------------------------------------- @ 36097/68174 “atoh” enhancer in human, mouse, rat, fugu fish
The local alignment problem Given two strings x = x1……xM, y = y1……yN Find substrings x’, y’ whose similarity (optimal global alignment value) is maximum e.g. x = aaaacccccgggg y = cccgggaaccaacc
Smith-Waterman Algorithm • Два отличия от Нидлмана-Вунша • Для каждого элемента матрицы дана возможность принять значение, равное нулю, если все другие значения отрицательны • Выравнивание может заканчиваться в любом месте таблицы. Лучший вес - наибольшее значение всей матрицы. Оттуда и начинается обратный проход 0 V(i-1, j)+d V(i, j) = max V(i, j-1)+d V(i-1, j-1)+score(si, tj)
empty G A T C A C C T 0 0 0 0 0 0 0 0 empty 0 0 0 0 0 G 0 0 0 0 0 A 0 0 0 0 1 T 0 0 0 0 0 A 0 0 C 0 0 0 0 C 0 0 0 C Local Alignment GATCACCT GATACCC GATCACCT GAT_ACCC • Let gap = -2 match = 1 mismatch = -1. 0 1 0 0 0 0 2 0 0 1 0 3 1 0 1 1 2 2 0 0 2 1 3 1 0 1 1 2 4 2 1 0 2 3 3
Overlap Alignment Перекрывающиеся выравнивания Consider the following problem: • Find the most significant overlap between two sequences S,T ? • Possible overlap relations: a. b. Difference from local alignment: Here we require alignment between the endpoints of the two sequences. Мы хотим получить разновидность глобального выравнивания, но в котором нет штрафа за свисающие концы То есть выравнивание начиналось на левой или верхней границе матрицы, а заканчивалось на правой или нижней
Overlap Alignment Формально: Исходя из S[1..n] , T[1..m] найти i,j такие что d - максимально, где d: d=max{D(S[1..i],T[j..m]) , D(S[i..n],T[1..j]) , D(S[1..n],T[i..j]) , D(S[i..j],T[1..m]) } . Решение: То же самое, что и глобальное выравнивание, за исключением того, что мы не штрафуем за висящие концы.
local overlap global Overlap Alignment • Initialization:V[i,0]=0,V[0,j]=0 Recurrence:as in global alignment Score:maximum value at the bottom line and rightmost line
Overlap Alignment (Example) S =PAWHEAE T =HEAGAWGHEE Scoring scheme : • Match: +4 • Mismatch: -1 • Indel: -5
Overlap Alignment (Example) S =PAWHEAE T =HEAGAWGHEE Scoring scheme : • Match: +4 • Mismatch: -1 • Indel: -5
Overlap Alignment (Example) S =PAWHEAE T =HEAGAWGHEE Scoring scheme: • Match: +4 • Mismatch: -1 • Indel: -5
Scoring scheme : • Match: +4 • Mismatch: -1 • Indel: -5 -2 Overlap Alignment (Example) The best overlap is: PAWHEAE------ ---HEAGAWGHEE Pay attention! A different scoring scheme could yield a different result, such as: ---PAW-HEAE HEAGAWGHEE-
Динамическое программирование с более сложными моделями • До сих пор мы рассматривали простейшую модель разрывов, где штраф d - линейно зависел от его длины. Каждый следующий остаток наказывается так же, как и первый. (g)= - nd n - число остатков, d - штраф за открытие разрыва • Введем аффинную функцию. (n)= -d-(n-1)e n - число остатков, d - штраф за открытие разрыва, а e - штраф за его продолжение
Dynamic Programming for the Affine Gap Penalty Case to do in time, need 3 matrices instead of 1 IGAxi LGVyi best score given that x[i] is aligned to y[j] best score given that x[i] is aligned to a gap AIGAxi GVyi-- best score given that y[j] is aligned to a gap GAxi-- SLGVyi
Why Three Matrices Are Needed consider aligning the sequences WFP and FW using d= -4 (gap opening), e = -1 (gap extension) and the following values from the BLOSUM-62 substitution matrix: W F P WF FW 0 -5 -6 -7 -WF FW- -WFP FW-- F -5 1 1 -4 optimal alignment W -6 6 2 0 best alignment of these prefixes; to get optimal alignment, need to also remember S(F, W) = 1 S(W, W) = 11 S(F, F) = 6 S(W, P) = -4 S(F, P) = -4 • the matrix shows the highest-scoring partial alignment for each pair of prefixes
Global Alignment DP for the Affine Gap Penalty Case d+e e d+e e M Ix Iy M Ix Iy M Ix Iy M Ix Iy M Ix Iy
Global Alignment DP for the Affine Gap Penalty Case initialization d+e d+e • traceback • start at largest of • stop at any of • note that pointers may traverse all three matrices
Global Alignment Example C A C T A M : 0 -∞ -∞ -∞ -∞ -∞ Ix : -3 -∞ -∞ -∞ -∞ -∞ Iy : -3 -4 -5 -6 -7 -8 -∞ 1 -5 -4 -7 -8 -4 -∞ -∞ -∞ -∞ -∞ A -∞ -∞ -3 -4 -5 -6 -∞ -3 0 -2 -5 -6 A -5 -3 -9 -8 -11 -12 -∞ -∞ -7 -4 -5 -6 -∞ -6 -4 -1 -3 -4 -6 -4 -4 -6 -9 -10 T -∞ -∞ -10 -8 -5 -6 ACACT AA--T ACACT A--AT ACACT --AAT three optimal alignments:
Local Alignment DP for the Affine Gap Penalty Case d+e e d+e e
Local Alignment DP for the Affine Gap Penalty Case initialization • traceback • start at largest • stop at