Download

1 / 6
60 likes | 70 Vues
As the development of biological experimental technology, especially gene-sequencing technology, both laboratory and clinic researchers realize that genome sequencing is the best way to analyze the etiology, pathophysiology, treatment and prognosis of diseases. Researches further demonstrate that<br>there are only 30 million base pairs of genes that contain essential information of proteins for human beings.

E N D
45-1 Ramsey Road Shirley, NY 11967 USA Email: contact@cd-genomics.com Principles and Workflow of Whole Exome Sequencing As the development of biological experimental technology, especially gene-sequencing technology, both laboratory and clinic researchers realize that genome sequencing is the best way to analyze the etiology, pathophysiology, treatment and prognosis of diseases. Researches further demonstrate that there are only 30 million base pairs of genes that contain essential information of proteins for human beings. The exome is ususally defined as the sequence encompassing all exons of protein coding genes, as well as nonprotein coding elements such as microRNA or lncRNA. The investigation of exome helps to figure out which loci are responsible for proper diseases. When researchers plan to explore exons information of human genome, the cost to whole genome sequencing will be quite surprising considering the total length of human genome is over 3 billion base pairs in size. To study rare mendelian diseases, exome sequencing is a more effective way to identify the genetic variants. The breakthrough of target-enrichment strategies and DNA sequencing techniques contributes to the development of whole exome sequencing. Principle of exome sequencing Exome sequencing contains two main processes, namely target-enrichment and sequencing. Target-enrichment is to select and capture exome from DNA samples. There are two major methods to achieve the enrichment of exome. ⚫Array-based exome enrichment uses probes bound to high-density microarrays to capture exome. A microarray is a 2 dimensional array on glass slide or silicon thin-film, which contains oligonucleotides complementary to target genome parts. While the fragmented DNA samples flow through microarray, the complementary pairing effect will force exome binding at microarray, with the other parts of genome remain dissociative, which results in the separation of exome from other parts of genome. ⚫In-solute capturing is based on magnetic bead. Magnetic bead is a kind of magnetic nanoparticles which contain functional chemical components to combine target substances. In this case, magnetic beads which could bind exome are used. Then the story is just the same with array-based method, exome is attracted and bound to the magnetic beads, with other parts of genome remain dissociative. The advantage of in-solute capturing method is that the usage of magnetic bead allows the reaction to be more effective by shaking or heating the system. Both of the methods are effective ways to extract exome from genome. So we say the sensitivities of both are high enough. However, the problem is specificity. There are parts of genome which share the similar sequence of some exons. Those parts of genome may bind to microarray or magnetic beads, resulting in false positive. Sequencing is the process to figure out the arrangement of all the deoxyribonucleotides in exome, which may help us to understand the potential pathophysiology alternation in some diseases. Because of the decrease of the cost, the importance of whole exome sequencing is prominent. The cost of human genome is approximately equal to two or three times the cost of whole exome sequencing. So
45-1 Ramsey Road Shirley, NY 11967 USA Email: contact@cd-genomics.com why not run more samples using whole exome sequencing to obtain more statistically significant result? General workflow of exome sequencing Here a common workflow of exome sequencing is shown as below. The instructions of the major processes in the workflow will be discussed below. Figure1. Workflow of whole exome sequencing. Notice that the detailed procedures are various from different types of samples, reagent kits and sequencing instruments. Researchers should follow the instructions of reagent, kits and sequencing instruments. ⚫Prepare your DNA samples: DNA fragmentation Almost all the experiment on DNA begins with DNA fragmentation. DNA should be sheared into proper pieces, because usually the length of DNA sample extracted from tissues or cells is too long. This shearing process is called DNA fragmentation. Effective target length is determined by the sequencing instrument that you choose. In order to process whole exome sequencing, there are several major ways to fragmentize DNA samples. Physical fragmentation. Physical fragmentation includes acoustic shearing, sonication and hydrodynamic shear. Among them acoustic shearing and sonication are the main methods for DNA fragmentation. DNA samples are broken into several pieces due to the acoustic cavitation and hydrodynamic shearing when they are exposed to ultrasound.
45-1 Ramsey Road Shirley, NY 11967 USA Email: contact@cd-genomics.com Enzymatic Methods. Enzymes used to break DNA into small pieces include nuclease or transposase. Nuclease will cleave the phosphodiester bonds between nucleic acids, resulting in the breaking down of DNA. Specifically, restriction endonuclease will cleave DNA at restriction sites. Transposase is used to mediate transposition events, processes that a certain DNA segment could "move around" the chromosome. It also plays a role in DNA fragmentation if we prepare appropriate DNA samples and transposase. The fragmentized DNA is linked with adapters instead of inserting again, resulting in fragmentation. After fragmentation, your DNA samples are ready for target-enrichment process. ⚫Isolation of exome: target-enrichment methods Exome has to be isolated from human genome before sequencing as the former contributes to only 1% of the latter. The process of capturing the target genomic regions is called target-enrichment. The basic idea of target-enrichment is to separate anything of interest from other substances using the physicochemical property difference between them. There are some common kits of target-enrichment methods. No matter what kit you choose, the variability in capture influence your exome sequencing, so be aware to the quality, quantity and fragment sizes of your DNA samples. Table1. Common kits of target-enrichment for sequencing. Kits Targeted Region 60 Mb 51 Mb 39 Mb 62 Mb Genomic DNA Input Required 100 ng 100 ng 500 ng 50 ng Adapter Addition Ligation Ligation Ligation Transposase Probe Length (mer) 120 120 not described 95 Agilent SureSelect XT2 V6 Exome Agilent SureSelect XT2 V5 Exome IDT xGEN Exome Panel Illumina Nextera Rapid Capture Expanded Exome Roche NimblegenSeqCap EZ Exome v3.0 64 Mb 1 ug Ligation 60 - 90 ⚫Harvest your products: washing and elution After the separation of exome and other parts of genome, several times of washing are required. The process of washing is just like what this word means literally -- to wash out anything we do not want so as to keep the thing of interest. In this case, we do not want substances such as the other parts of genome, proteins, and electrolytes. Distilled water is usually used to elute target, but some special reagent kits may require specific eluent. Eluent is the reagent to wash down the exome from microarray or magnetic beads, which is able to break the connection between exome and binding substances. Both washing and elution process could be processed multiple times in order to obtain purer exomes. Also in some cases, one more target-enrichment process is performed to make the elution better. Just follow the instruction of reagent kit you used, and adjust your protocol according to your actual situation. ⚫Sequencing technology Because of time cost and length requirement of the Sanger Sequencing, the sequencing technology did not contribute much in biological and clinical studies, until next generation sequencing (NGS) technologies are invented. NGS technologies are based on the usage of dyed ddNTPs in Sanger method. The improvement is that NGS allows DNA strands to be combined, amplified and detected at
45-1 Ramsey Road Shirley, NY 11967 USA Email: contact@cd-genomics.com the same time, leading to breakneck increase in length requirement and efficiency of sequencing. To simplify, the principle of NGS is to bind the exome samples in a proper base (such as flowcell of Illumina Hiseq and magnetic beads of Roche-454) and replicate them by PCR-in-situ, in order to make signal in every rounds of elongation amplified. Then ddNTPs are detected after every round of elongation. Finally, the complete sequence is integrated using biological information algorithm. NGS largely improves the efficiency and allows higher-throughput detection, that is why NGS is also called high-throughput sequencing and is widely used. Besides of NGS, the third generation of sequencing is developing rapidly, which largely exceeds the efficiency of NGS. The key feature of third generation sequencing is single-molecule sequencing. It shortens the time cost of whole genome sequencing to several minutes. Companies such as PacificBio and Oxford Nanopore have proved their method works, and third generation of sequencing technology could lead a revolution in exome sequencing area. Here are some common methods of whole exome sequencing used nowadays. Table2. Common methods used for sequencing nowadays. Methods Company Generation Accuracy Reads per run Time per run Read length Up to 600 bp 700 bp Ion semiconductor Pyrosequencing (454) Sequencing by synthesis Sequencing by ligation (SOLiD) Nanopore Sequencing Ion Torrent 2nd generation 2nd generation 2nd generation 2nd generation 3rd generation 99.60% up to 80 million 2 hours Roche 99.90% 1 million 24 hours Illumina 75-300 bp 99.90% 1 million to 3 billion 1.2 to 1.4 billion 1 to 2 weeks 1 to 11 days ABI 50+35 or 50+50 bp up to 500 kb 99.90% Oxford Nanopore Technologies 92–97% (single read)* dependent on read length selected by user 10-20 billion 1 min to 48 hours Single-molecule real-time sequencing *For third generation sequencing, accuracy is usually improved by sequencing for multiple times. Pacific Biosciences 3rd generation 30,000 bp 87% (single read)* 0.5-20 hours ⚫Data analysis The data of sequencing are confusing and unreadable before bioinformatics analysis and interpretation, because most of the sequencing methods produce short fragments of sequence, which require sequence assembly to figure out the final result. The following pipeline can be used by researchers who are interested in performing WES analysis for variant calling and genetic diseases.
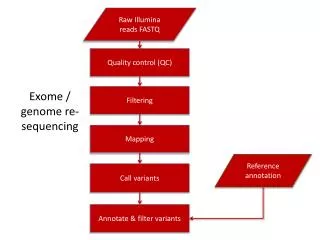
45-1 Ramsey Road Shirley, NY 11967 USA Email: contact@cd-genomics.com Figure2. The typical variant calling pipeline. Conclusion We have benefited a lot from exome sequencing in both academic research and clinic diagnosis. Thanks to exome sequencing, the understanding of genome is developed to a new level. Many diseases used to be mysteries, such as neurological disorder in infants, which could be predicted now. Furthermore, many diseases with few treatments, such as carcinoma, are allowed to be treated by targeted therapy. It is said that the fourth generation of sequencing technology is developing. Hope it would drive another revolution in biological and medical research. References: 1. Teer JK and Mullikin JC (2010) 'Exome sequencing: the sweet spot before whole genomes ', Hum Mol Genet, 19(R2), R145-51. 2. Amanda Warr, Christelle Robert, David Hume, Alan Archibald, Nader Deeb and Mick Watson (2015) 'Exome Sequencing: Current and Future Perspectives', G3 (Bethesda), 5(8), 1543–1550. 3. Phillips and Thearesa. (2013) 'Restriction Enzymes Explained', G3 (Bethesda), 5(8), 1543–1550. 4. Stavros Basiardes; Rose Veile; Cindy Helms; Elaine R. Mardis; Anne M. Bowcock; Michael Lovett (2005) 'Direct Genomic Selection', Nature Methods, 1 (2), 63–69. 5. Tadic, Marin; Kralj, Slavko; Jagodic, Marko; Hanzel, Darko; Makovec and Darko (2014). 'Magnetic properties of novel superparamagnetic iron oxide nanoclusters and their peculiarity under annealing treatment', Applied Surface Science, 322, 255–264. 6. Sanger F and Coulson AR (1975). 'A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase', J. Mol. Biol, 94 (3), 441–8. 7. Van Vliet AH (2010). Next generation sequencing of microbial transcriptomes: challenges and opportunities. '', FEMS Microbiol Lett. 302(1), 1-7.
45-1 Ramsey Road Shirley, NY 11967 USA Email: contact@cd-genomics.com 8. Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, Turner SW, Korlach J (2013). "Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data". Nat.Methods. 10 (6), 563–69. 9. Nolan D and Carlson M (2016). "Whole Exome Sequencing in Pediatric Neurology Patients: Clinical Implications and Estimated Cost Analysis."J Child Neurol. 31(7), 887-94.. 10. Salazar-García L, Pérez-Sayáns M, García-García A, Carracedo A, Cruz R3, Lozano A, Sobrino B and Barros F. "Whole exome sequencing approach to analysis of the origin of cancer stem cells in patients with head and neck squamous cell carcinoma."J Oral Pathol Med. doi: 10.1111/jop.12771.