
The US Food and Drug Administration plays a central role in ensuring the safety and effectiveness of medical devices used in healthcare. When the FDA announces approved or cleared medical devices, it also provides important risk-related information that helps manufacturers, healthcare professionals, and patients understand how these devices should be used safely. FDA risk information is not limited to the premarket stage; it continues throughout the entire lifecycle of a medical device and supports ongoing patient protection.
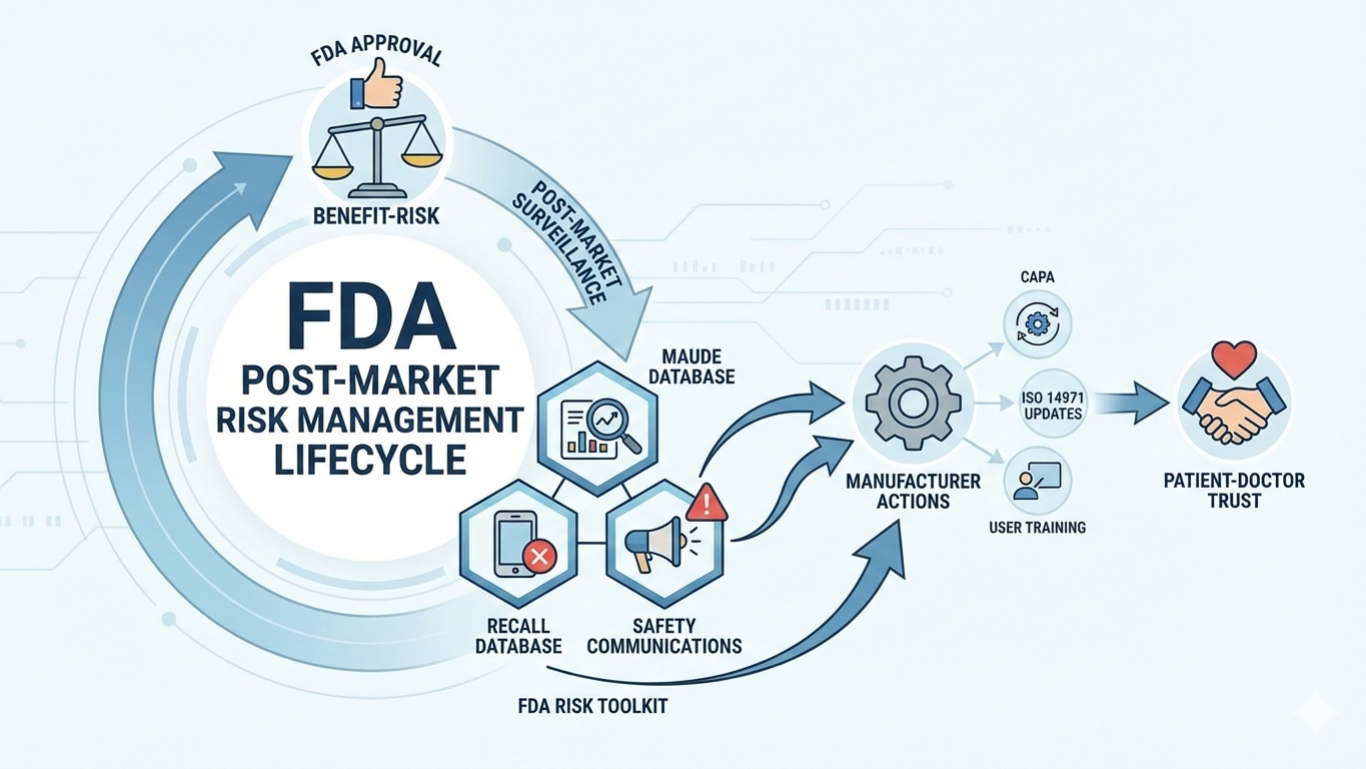
FDA approval or clearance means that a medical device has met specific regulatory requirements based on its risk classification and intended use. Devices are categorized into Class I, Class II, or Class III depending on the level of risk they pose. Lower-risk devices generally undergo fewer regulatory controls, while higher-risk devices require more extensive review and clinical evidence. Approval or clearance confirms that the FDA has evaluated available data and determined that the benefits of the device outweigh its potential risks when used as intended. However, this does not eliminate the need for continuous risk monitoring after the device enters the market. FDA risk information is built on a structured risk–benefit approach. During review, the FDA evaluates potential hazards, clinical performance, failure modes, and mitigation measures. This information helps determine whether risks can be adequately controlled through design, labeling, instructions for use, or post-market controls. Even after market entry, the FDA continues to assess real-world device performance to identify emerging safety concerns that may not have been evident during premarket evaluation. Post-market surveillance is a key component of FDA risk management.
The FDA collects and analyzes safety data through multiple systems, including adverse event reports, recall data, and safety communications. One of the most important tools is the MAUDE database, which captures reports from manufacturers, healthcare facilities, and users about device malfunctions, injuries, or deaths. This data allows regulators and manufacturers to detect trends, investigate potential safety issues, and take corrective actions when necessary. Risk information published by the FDA is particularly valuable for medical device manufacturers. It supports ongoing risk management activities, including design improvements, corrective and preventive actions, and updates to technical documentation. Manufacturers are required to maintain post-market vigilance, investigate complaints, and report serious adverse events within defined timelines. Integrating FDA risk data into quality management systems helps ensure compliance with regulatory requirements and supports continuous product improvement. Healthcare providers and patients also benefit from transparent FDA risk information. Access to recall notices, safety alerts, and device performance updates enables informed decision-making and safer clinical use.
Clear communication of risks supports proper device selection, correct usage, and timely response to safety issues. This transparency strengthens trust in the regulatory system and promotes patient safety across healthcare settings. After FDA approval, regulatory responsibilities continue for manufacturers. Ongoing post-market surveillance, periodic reporting, and risk management reviews are essential to maintaining compliance. Risk management is a lifecycle process, not a one-time regulatory task. Manufacturers must actively monitor device performance, respond to new risk information, and implement changes when necessary to protect patients and users.
Understanding FDA risk information for approved medical devices is essential for navigating regulatory expectations and ensuring long-term compliance. By actively using FDA safety data, manufacturers and healthcare stakeholders can enhance patient protection, improve device quality, and maintain confidence in regulated medical technologies. For additional regulatory insights on FDA risk information and post-market requirements, educational resources from platforms such as Operon Strategist provide structured guidance on aligning risk management practices with FDA expectations.







