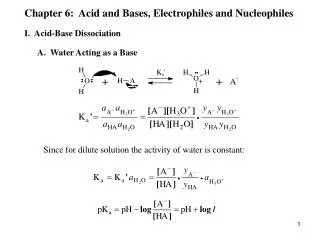
Lone Pair acting as Base.
Lone Pair acting as Base. Note the change in formal charges. As reactant oxygen had complete ownership of lone pair. In product it is shared. Oxygen more positive by 1. Similarly, B has gained half of a bonding pair; more negative by 1. An example: pi electrons as bases. Bronsted Lowry Base.
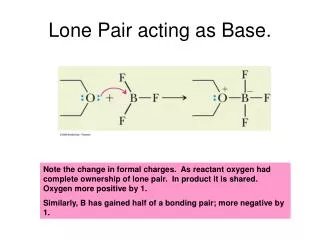

Lone Pair acting as Base.
E N D
Presentation Transcript
Lone Pair acting as Base. Note the change in formal charges. As reactant oxygen had complete ownership of lone pair. In product it is shared. Oxygen more positive by 1. Similarly, B has gained half of a bonding pair; more negative by 1.
An example: pi electrons as bases Bronsted Lowry Base Bronsted Lowry Acid For the moment, just note that there are two possible carbocations formed. The carbocations are conjugate acids of the alkenes.
Sigma bonding electrons as bases. Much more unusual!! A very, very electronegative F!! A very positive S!! The OH becomes very acidic because that would put a negative charge adjacent to the S. Super acid
Trends for Relative Acid Strengths Totally unionized in aqueous solution Aqueous Solution Totally ionized in aqueous solution.
Example pKa = 15.9 Weaker acid pKa = 9.95 Stronger acid H2O + PhOH H3O+ + PhO- H2O + EtOH H3O+ + EtO- Ka = [H3O+][EtO-]/[EtOH] = 10-15.9 Ka = [H3O+][PhO-]/[PhOH] = 10-9.95 Ethanol, EtOH, is a weaker acid than phenol, PhOH. It follows that ethoxide, EtO-, is a stronger base than phenolate, PhO-. For reaction PhOH + EtO- PhO- + EtOH where does equilibrium lie? Weaker base. Stronger base K = 10-9.95 /10-15.9 = 106.0 Query: What makes for strong (or weak) acids?
What affects acidity? Increasing basicity of anion. Increasing basicity of anion. 1. Electronegativity of the atom holding the negative charge. Increasing electronegativity of atom bearing negative charge. Increasing stability of anion. Increasing acidity. 2. Size of the atom bearing the negative charge in the anion. Increasing acidity. Increasing size of atom holding negative charge. Increasing stability of anion.
What affects acidity? - 2 3. Resonance stabilization, usually of the anion. Increasing resonance stabilization. Increased anion stability. Increasing basicity of the anion. Acidity No resonance structures!! Note that phenol itself enjoys resonance but charges are generated, costing energy, making the resonance less important. The more important resonance in the anion shifts the equilibrium to the right making phenol more acidic.
Two different bases or two sites in the same molecule may compete to be protonated (be the base). An example: competitive Bases & Resonance Acetic acid can be protonated at two sites. Pi bonding electrons converted to non-bonding. Which conjugate acid is favored? The more stable one! Which is that? Recall resonance provides additional stability by moving pi or non-bonding electrons. No valid resonance structures for this cation. Non-bonding electrons converted to pi bonding.
An example: competitive Bases & Resonance Comments on the importance of the resonance structures. All atoms obey octet rule! The carbon is electron deficient – 6 electrons, not 8. Lesser importance All atoms obey octet rule!
What affects acidity? - 3 4. Inductive and Electrostatic Stabilization. Increasing anion stability. Increasing anion basicity. Acidity. d+ d+ Due to electronegativity of F small positive charges build up on C resulting in stabilization of the anion. Effect drops off with distance. EtOH pKa = 15.9
What affects acidity? - 4 Note. The NH2- is more basic than the RCC- ion. 5. Hybridization of the atom bearing the charge. H-A H+ + A:-. sp3 sp2 sp More s character, more stability, more “electronegative”, H-A more acidic, A:- less basic. Increasing Acidity of HA Increasing Basicity of A- Know this order.
What affects acidity? - 5 6. Stabilization of ions by solvents (solvation). Solvation provides stabilization. Comparison of alcohol acidities. 17 18 pKa = 15.9 Crowding inhibiting solvation (CH3)3CO -, crowded Solvation, stability of anion, acidity
Example Para nitrophenol is more acidic than phenol. Offer an explanation The lower lies further to the right. Why? Could be due to destabilization of the unionized form, A, or stabilization of the ionized form, B. B A
Examine the equilibrium for p-nitrophenol. How does the nitro group increase the acidity? Examine both sides of equilibrium. What does the nitro group do? First the unionized acid. Note carefully that in these resonance structures charge is created: + on the O and – in the ring or on an oxygen. This decreases the importance of the resonance. Structure D occurs only due to the nitro group. The stability it provides will slightly decrease acidity. Resonance structures A, B and C are comparable to those in the phenol itself and thus would not be expected to affect acidity. But note the + to – attraction here
Now look at the anion. What does the nitro group do? Remember we are interested to compare with the phenol phenolate equilibrium. In these resonance structures charge is not created. Thus these structures are important and increase acidity. They account for the acidity of all phenols. Structure D occurs only due to the nitro group. It increases acidity. The greater amount of significant resonance in the anion accounts for the nitro increasing the acidity. Resonance structures A, B and C are comparable to those in the phenolate anion itself and thus would not be expected to affect acidity. But note the + to – attraction here
Shape All atoms in same plane except for these hydrogens on sp3 carbon. Alkenes, 6 coplanar atoms.
Arene shapes Planar ring structure. 12 atoms coplanar. Phenyl group, C6H5,, Ph Ph = C6H5
Pi bonds Energy
Nomenclature Z / E generalization of cis / trans Use R, S priorities to compare substituents on same carbon. High priority on same side, Z. Opposite, E.
Cis / Trans in Cycloalkenes For small rings normally have cis double bonds.
Terpenes and the isoprene Rule A terpene is composed of isoprene units joined head to tail (the isoprene rule). This moleculehas additional cross links. Note that location of functional groups such as OH or double bonds is not addressed.
Vitamin A Four isprene units joined head to tail One cross link (non-head to tail) linkage.
Fatty Acids Animal fats and vegetable oils are both triesters of glycerol, hence the name triglyceride. Hydrolysis of a triglyceride in aqueous base followed by acidification gives glycerol and three fatty acids. Fatty acids with no C=C double bonds are called saturated fatty acid. Those with one or more C=C double bonds are called unsaturated fatty acids.
Fatty Acids The most common fatty acids have an even number of carbons, and between 12 and 20 carbons in an unbranched chain. The C=C double bonds in almost all naturally occurring fatty acids have a cis configuration. The greater degree of unsaturation, the lower the melting point. Triglycerides rich in unsaturated fatty acids are generally liquid at room temperature and are called oils. Triglycerides rich in saturated fatty acids are generally semisolids or solids at room temperature and are called fats.
Fatty Acids the four most abundant fatty acids
Pi bonds Reactivity above and below the molecular plane! Plane of molecule
Addition Reactions • Important characteristics of addition reactions • Orientation (Regioselectivity) • If the doubly bonded carbons are not equivalent which one get the A and which gets the B. • Stereochemistry: geometry of the addition. • Syn addition: Both A and B come in from the same side of the alkene. Both from the top or both from the bottom. • Anti Addition: A and B come in from opposite sides (anti addition). • No preference.
Reaction Mechanisms Mechanism: a detailed, step-by-step description of how a reaction occurs. A reaction may consist of many sequential steps. Each step involves a transformation of the structure. For the step C + A-B C-A + B Transition State Three areas to be aware of. Products Reactants Energy of Activation. Energy barrier.
Energy Changes in a Reaction Enthalpy changes, DH0, for a reaction arises from changes in bonding in the molecule. If weaker bonds are broken and stronger ones formed then DH0 is negative and exothermic. If stronger bonds are broken and weaker ones formed then DH0 is positive and endothermic.
Gibbs Free Energy Gibbs Free Energy controls the position of equilibrium for a reaction. It takes into account enthalpy, H, and entropy, S, changes. An increase in H during a reaction favors reactants. A decrease favors products. An increase in entropy (eg., more molecules being formed) during a reaction favors products. A decrease favors reactants. DG0: if positive equilibrium favors reactants (endergonic), if negative favors products (exergonic). DG0 = DH0 – TDS0
Multi-Step Reactions Step 1 is the “slow step”, the rate determining step. Step 1: A + B Intermediate Step 2: Intermediate C + D Step 2: exergonic, small energy of activation. Fast Process. Step 1: endergonic, high energy of activation. Slow process
Characteristics of two step Reaction • The Intermediate has some stability. It resides in a valley. • The concentration of an intermediate is usually quite low. The Energies of Activation for reaction of the Intermediate are low. • There is a transition state for each step. A transition state is not a stable structure. • The reaction coordinate can be traversed in either direction: A+B C+D or C+D A+B.
Hammond Postulate The transition state for a step is close to the high energy end of the curve. For an endothermic step the transition state resembles the product of the step more than the reactants. For an exothermic step the transition state resembles the reactants more than the products. Reaction coordinate.
Example • Endothermic • Transition state resembles the (higher energy) products. Almost formed radical. Only a small amount of radical character remains. Almost formed. Almost broken.
Electrophilic Additions Hydrohalogenation using HCl, HBr, HI Hydration using H2O in the presence of H2SO4 Halogenation using Cl2, Br2 Halohydrination using HOCl, HOBr Oxymercuration using Hg(OAc)2, H2O followed by reduction
Electrophilic Addition We now address regioselectivity….
Regioselectivity (Orientation) The incoming hydrogen attaches to the carbon with the greater number of hydrogens. This is regioselectivity. It is called Markovnikov orientation.
Mechanism Step 1 Step 2
Now examine Step 1 Closely Electron rich, pi system. Showed this reaction earlier as an acid/base reaction. Alkene was the base. New term: the alkene is a nucleophile, wanting to react with a positive species. Acidic molecule, easily ionized. We had portrayed the HBr earlier as a Bronsted-Lowry acid. New term: the HBr is an electrophile, wanting to react with an electron rich molecule (nucleophile). Rate Determining Step. The rate at which the carbocation is formed controls the rate of the overall reaction. The energy of activation for this process is critical. The carbocation intermediate is very reactive. It does not obey the octet rule (electron deficient) and is usually present only in low concentration.
Carbocations sp2 hybridized. p orbital is empty and can receive electrons. Flat, planar. Can react on either side of the plane. Very reactive and present only in very low concentration. Electron deficient. Does not obey octet rule. Lewis acid, can receive electrons. Electrophile.
Step 2 of the Mechanism :Br- Mirror objects :Br-
Regioselectivity (Orientation) Or Primary carbocation Secondary carbocation Secondary carbocation more more stable and more easily formed.
Carbocation Stabilities Order of increasing stability: Methyl < Primary < Secondary < Tertiary Order of increasing ease of formation: Methyl < Primary < Secondary < Tertiary Increasing Ease of Formation
Factors Affecting Carbocation Stability - Inductive • Inductive Effect. Electron redistribution due to differences in electronegativities of substituents. • Electron releasing, alkyl groups, -CH3, stabilize the carbocation making it easier to form. • Electron withdrawing groups, such as -CF3, destabilize the carbocation making it harder to form. d- d+ d- d-
Factors Affecting Carbocation Stability - Hyperconjugation 2. Hyperconjugation. Unlike normal resonance or conjugation hyperconjugation involves s bonds. Hyperconjugation spreads the positive charge onto the adjacent alkyl group