Download

1 / 33
340 likes | 1.15k Vues
Finding identities and similarities in nucleotide and protein sequences: What proteins are like mine? What the evolutionary history? Can I infer function of my gene/protein?. Homework is at the end!!!!. Overview. Similarity vs homology Orthologs and paralogs

E N D
Finding identities and similarities in nucleotide and protein sequences:What proteins are like mine?What the evolutionary history?Can I infer function of my gene/protein? Homework is at the end!!!!
Overview • Similarity vs homology • Orthologs and paralogs • Global vs local sequencing alignments • Scoring matrices • BLAST • Other search programs
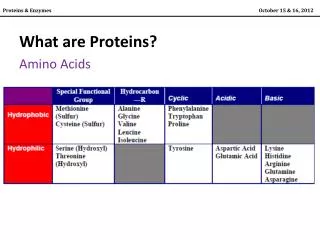
Terminology • Similarity (percent identity), measures sequence divergence • Homology – evolutionary relatedness (are or are not homologous) • Orthologs are genes separated by a speciation event • Paralogs are genes separated by a duplication event • Identity the extent that a pair or more sequences are related by comparison (either by same base or residue at a equivalent position expressed as a percent).
Why do alignments? • A proved measure of relatedness between nucleotide or amino acid sequences • With this it is possible to infer biological relationships • Structural, functional, evolutionary
Global vs. Local Alignment • Global alignments attempt to optimally align the entire length of two sequences. They are often used to compare sequences thought to be somewhat similar. As sequence similarity declines, this may miss important biological relationships. • Local alignments look for regions of local sequence simliarity. They are often used to evaluate sequences thought to be somewhat dissimilar. • http://www.ncbi.nlm.nih.gov/blast/producttable.shtml#blastx
Basic matrix example Align the sequences ACCATTGCC and ATTG using a simple matrix Assign all matches in a sequence comparison a value Example: 1 for a match and 0 for a mismatch (you have to decide how much you want to penalize a mismatch). A T T G C 0 0 0 0 C 0 0 0 0 A 1 0 0 0 T 0 1 0 0 T 0 0 1 0 G 0 0 0 1 C 0 0 0 0 C 0 0 0 0 Basic presentation of matrices: http://biology.unm.edu/biology/maggieww/Public_Html/matrixdiscussion.htm Great BLAST tutorial: http://genomebiology.com/content/pdf/gb-2001-2-10-reviews2002.pdf
BLAST • The Basic Local Alignment Search Tool (BLAST) finds regions of local similarity between sequences. • The program (algorithm) compares nucleotide or protein sequences to sequences in databases • BLAST calculates the statistical significance of matches based on expectations, assuming independence of each nucleotide or amino acid. Expectation is also a function of the size of the database being compared. • Most commonly used method http://blast.ncbi.nlm.nih.gov/Blast.cgi
How to do your search? • Which BLAST program is appropriate? • nucleotide blast Search a nucleotide database using a nucleotide queryAlgorithms: blastn, megablast, discontiguous megablast • protein blast Search protein database using a protein queryAlgorithms: blastp, psi-blast, phi-blast • blastx Search protein database using a translated nucleotide query • tblastn Search translated nucleotide database using a protein query tblastx Search translated nucleotide database using a translated nucleotide query http://blast.ncbi.nlm.nih.gov/Blast.cgi
BLASTn example ATCGGACGTGGATCCATCGATCGATGCGATCGATCGAAATCG sequence that you want to know about
BLASTn example Which Database or organism would you like to search
BLASTn example ATCGGACGTGGATCCATCGATCGATGCGATCGATCGAAATCG Search type and stringency
General parameters • For Short Queries if you check the box, it will automatically adjust the word size and other parameters to improve results for short query sequence lengths. • Word Size Allows you to change the length of the seed that initiates an alignment.
Expect threshold • This setting specifies the statistical significance threshold for reporting matches against database sequences. • The default value (10) means that 10 such matches are expected to be found merely by chance • A match is greater than the EXPECT threshold, the match will not be reported. Lower EXPECT thresholds are more stringent, leading to fewer chance matches being reported.
Scoring parameters • Many nucleotide searches use a simple scoring system that consists of a "reward" for a match and a "penalty" for a mismatch. • This ratio should be increased as one looks at more divergent sequences. • Example • (1/-3) for sequences that are about 99% conserved • (1/-2) is best for sequences that are 95% conserved • (1/-1) is best for sequences that are 75% conserved
Filters and masking • Filters allow the program to ignore areas of the genome with a high number of repeats or low complexity from the alignment. Otherwise, including these regions could produce to misleading results. • Masking allows for alterations in the input data such as masking lowercase as uppercase letters and modifying other features common in FASTA format
PAM vs. BLOSUM for general users • The larger PAM (Point Accepted Mutations ) matrix numbers are used to evaluate sequences with greater dissimilarity or distance. (PAM150 is used for more distant sequences than PAM100) based on global alignments • The larger BLOSUM (BLOcksSUbstitution Matrix ) matrix numbers are used to evaluate sequences with higher sequence similarity or less distance. (BLOSUM62 is used for closer sequences than Blosum50.) based on local alignments http://blast.ncbi.nlm.nih.gov/Blast.cgi PAM ref paper: Dayhoff, M.O., Schwartz, R. and Orcutt, B.C. (1978). "A model of Evolutionary Change in Proteins". Atlas of protein sequence and structure (volume 5, supplement 3 ed.). Nat. Biomed. Res. Found.. p. 345–358. BLOSUM ref paper: Henikoff, S.; Henikoff, J.G. (1992). "Amino Acid Substitution Matrices from Protein Blocks". PNAS89 (22): 10915–10919.
PAM vs. BLOSUM for general users http://www.google.com/imgres?imgurl=http://www.clcbio.com/scienceimages/pam-blosum.png
Alignment score • A summation of each specified aligned pair of bases or residues, and their nulls, in the alignment. The higher the alignment score, the better the alignment. http://blast.ncbi.nlm.nih.gov/Blast.cgi
Max and Total score • high scoring pairs = HSPs • Max Score: The higher the Max Score, the better the alignment between the hit and the query. This is based on the overall score of HSPs between sequences, similar to Expect Value • Total Score: By the sum of scores from all HSPs from the same database sequence. http://blast.ncbi.nlm.nih.gov/Blast.cgi
E (expected) Value • it describes the chance of randomly achieving the same alignment in a database of a particular size. • An E Value is used to describe the significance (instead of a P value) of each sequence alignment hit to the query. • The lower the E value is, the more significant the alignment is. http://blast.ncbi.nlm.nih.gov/Blast.cgi
Query Coverage and Max Identity • The amount of the query sequence, expressed as a percent, that overlaps the subject sequence • the highest percent identity for a set of aligned segments to the same subject sequence. http://blast.ncbi.nlm.nih.gov/Blast.cgi
Blastp, PSI-BLAST, PHI-BLAST BlastP simply compares a protein query to a protein database. PSI-BLAST allows the user to build a PSSM (position-specific scoring matrix) using the results of the first BlastP run.) PHI-BLAST performs the search but limits alignments to those that match a pattern in the query.
Homework Questions (sequences on subsequent slides) • Go to NCBI BLAST, perform a ‘megablast’ BLASTn. First, use the ‘Human’ databases and then ‘Others’. What are the differences? How did the E-values, scores, identities, organisms, gene names and coverage change? What did you not expect about the results? • Use the tBLASTx and BLASTx on the translated sequence. For tBLASTx try BLOSUM80 and 45 scoring matrices. What are the differences? How did the E-values, scores, identities, organisms, gene names and coverage change? What do the hits with different BLOSUM scores tell you? What did you not expect about the results? • Compare the usefulness of your results to each other. What type of questions would you be better able to address depending on which algorithm and sequence type used for your queries (translated or nucleotide?) (hints: evolution, model organisms)
This is a FASTA DNA seq for BRCA1 • >gi|237757283|ref|NM_007294.3| Homo sapiens breast cancer 1, early onset (BRCA1), transcript variant 1, mRNA GTACCTTGATTTCGTATTCTGAGAGGCTGCTGCTTAGCGGTAGCCCCTTGGTTTCCGTGGCAACGGAAAA GCGCGGGAATTACAGATAAATTAAAACTGCGACTGCGCGGCGTGAGCTCGCTGAGACTTCCTGGACGGGG GACAGGCTGTGGGGTTTCTCAGATAACTGGGCCCCTGCGCTCAGGAGGCCTTCACCCTCTGCTCTGGGTA AAGTTCATTGGAACAGAAAGAAATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATTAAT GCTATGCAGAAAATCTTAGAGTGTCCCATCTGTCTGGAGTTGATCAAGGAACCTGTCTCCACAAAGTGTG ACCACATATTTTGCAAATTTTGCATGCTGAAACTTCTCAACCAGAAGAAAGGGCCTTCACAGTGTCCTTT ATGTAAGAATGATATAACCAAAAGGAGCCTACAAGAAAGTACGAGATTTAGTCAACTTGTTGAAGAGCTA TTGAAAATCATTTGTGCTTTTCAGCTTGACACAGGTTTGGAGTATGCAAACAGCTATAATTTTGCAAAAA AGGAAAATAACTCTCCTGAACATCTAAAAGATGAAGTTTCTATCATCCAAAGTATGGGCTACAGAAACCG TGCCAAAAGACTTCTACAGAGTGAACCCGAAAATCCTTCCTTGCAGGAAACCAGTCTCAGTGTCCAACTC TCTAACCTTGGAACTGTGAGAACTCTGAGGACAAAGCAGCGGATACAACCTCAAAAGACGTCTGTCTACA TTGAATTGGGATCTGATTCTTCTGAAGATACCGTTAATAAGGCAACTTATTGCAGTGTGGGAGATCAAGA ATTGTTACAAATCACCCCTCAAGGAACCAGGGATGAAATCAGTTTGGATTCTGCAAAAAAGGCTGCTTGT GAATTTTCTGAGACGGATGTAACAAATACTGAACATCATCAACCCAGTAATAATGATTTGAACACCACTG AGAAGCGTGCAGCTGAGAGGCATCCAGAAAAGTATCAGGGTAGTTCTGTTTCAAACTTGCATGTGGAGCC ATGTGGCACAAATACTCATGCCAGCTCATTACAGCATGAGAACAGCAGTTTATTACTCACTAAAGACAGA ATGAATGTAGAAAAGGCTGAATTCTGTAATAAAAGCAAACAGCCTGGCTTAGCAAGGAGCCAACATAACA GATGGGCTGGAAGTAAGGAAACATGTAATGATAGGCGGACTCCCAGCACAGAAAAAAAGGTAGATCTGAA TGCTGATCCCCTGTGTGAGAGAAAAGAATGGAATAAGCAGAAACTGCCATGCTCAGAGAATCCTAGAGAT ACTGAAGATGTTCCTTGGATAACACTAAATAGCAGCATTCAGAAAGTTAATGAGTGGTTTTCCAGAAGTG ATGAACTGTTAGGTTCTGATGACTCACATGATGGGGAGTCTGAATCAAATGCCAAAGTAGCTGATGTATT GGACGTTCTAAATGAGGTAGATGAATATTCTGGTTCTTCAGAGAAAATAGACTTACTGGCCAGTGATCCT
This is a FASTA Translated seq for BRCA1 • >BRCA1 translation="MDLSALRVEEVQNVINAMQKILECPICLELIKEPVSTKCDHIFC KFCMLKLLNQKKGPSQCPLCKNDITKRSLQESTRFSQLVEELLKIICAFQLDTGLEYA NSYNFAKKENNSPEHLKDEVSIIQSMGYRNRAKRLLQSEPENPSLQETSLSVQLSNLG TVRTLRTKQRIQPQKTSVYIELGSDSSEDTVNKATYCSVGDQELLQITPQGTRDEISL DSAKKAACEFSETDVTNTEHHQPSNNDLNTTEKRAAERHPEKYQGSSVSNLHVEPCGT NTHASSLQHENSSLLLTKDRMNVEKAEFCNKSKQPGLARSQHNRWAGSKETCNDRRTP STEKKVDLNADPLCERKEWNKQKLPCSENPRDTEDVPWITLNSSIQKVNEWFSRSDEL LGSDDSHDGESESNAKVADVLDVLNEVDEYSGSSEKIDLLASDPHEALICKSERVHSK SVESNIEDKIFGKTYRKKASLPNLSHVTENLIIGAFVTEPQIIQERPLTNKLKRKRRP TSGLHPEDFIKKADLAVQKTPEMINQGTNQTEQNGQVMNITNSGHENKTKGDSIQNEK NPNPIESLEKESAFKTKAEPISSSISNMELELNIHNSKAPKKNRLRRKSSTRHIHALE LVVSRNLSPPNCTELQIDSCSSSEEIKKKKYNQMPVRHSRNLQLMEGKEPATGAKKSN KPNEQTSKRHDSDTFPELKLTNAPGSFTKCSNTSELKEFVNPSLPREEKEEKLETVKV SNNAEDPKDLMLSGERVLQTERSVESSSISLVPGTDYGTQESISLLEVSTLGKAKTEP NKCVSQCAAFENPKGLIHGCSKDNRNDTEGFKYPLGHEVNHSRETSIEMEESELDAQY LQNTFKVSKRQSFAPFSNPGNAEEECATFSAHSGSLKKQSPKVTFECEQKEENQGKNE SNIKPVQTVNITAGFPVVGQKDKPVDNAKCSIKGGSRFCLSSQFRGNETGLITPNKHG LLQNPYRIPPLFPIKSFVKTKCKKNLLEENFEEHSMSPEREMGNENIPSTVSTISRNN IRENVFKEASSSNINEVGSSTNEVGSSINEIGSSDENIQAELGRNRGPKLNAMLRLGV LQPEVYKQSLPGSNCKHPEIKKQEYEEVVQTVNTDFSPYLISDNLEQPMGSSHASQVC