MUTAZIONI CROMOSOMICHE NELL’UOMO
450 likes | 2.12k Vues
MUTAZIONI CROMOSOMICHE NELL’UOMO. ANOMALIE NUMERICHE. EUPLOIDI DESCRITTE SOLO IN ABORTI SPONTANEI MOLTO PRECOCI TRIPLOIDIE, MOSAICI TRIPLOIDI, TETRAPLOIDIE, MOSAICI TETRAPLOIDI. ANEUPLOIDI ANEUPLOIDIE DEGLI AUTOSOMI POCHISSIME SONO COMPATIBILI CON LA VITA

MUTAZIONI CROMOSOMICHE NELL’UOMO
E N D
Presentation Transcript
ANOMALIE NUMERICHE EUPLOIDI • DESCRITTE SOLO IN ABORTI SPONTANEI MOLTO PRECOCI • TRIPLOIDIE, MOSAICI TRIPLOIDI, TETRAPLOIDIE, MOSAICI TETRAPLOIDI
ANEUPLOIDI • ANEUPLOIDIE DEGLI AUTOSOMI • POCHISSIME SONO COMPATIBILI CON LA VITA • MAI DESCRITTE MONOSOMIE NEI NATI VIVI • GRAVE RITARDO MENTALE E DI SVILUPPO • MALFORMAZIONI MULTIPLE SCHELETRICHE E DEGLI ORGANI INTERNI • FACIES CARATTERISTICA
ANEUPLOIDIE DEI CROMOSOMI DEL SESSO N.B.B.: INATTIVAZIONE DEL CROMOSOMA X, Y POVERO DI GENI • SONO LE MEGLIO TOLLERATE • SONO LE UNICHE MONOSOMIE NOTE • SONO ABBASTANZA FREQUENTI POLISOMIE X ED Y • LIEVE RITARDO MENTALE • BLANDE ANOMALIE SOMATICHE • ANOMALIE DI SVILUPPO • ANOMALIE NELLO SVILUPPO DEI CARATTERI SESSUALI SECONDARI • STERILITA'
SINDROME DI DOWN • RITARDO MENTALE CAUSATO DA UNA COPIA EXTRA DEL CROMOSOMA 21 • DESCRITTA PER LA PRIMA VOLTA DA LANGON DOWN NEL 1866 • SCOPERTA LA PRESENZA DI UN 21 IN PIU’ NEL 1959 (LEJEUNE)
SINDROME DI DOWN47,XX XY, 21+ FREQUENZA NEI NATI VIVI 1/780 (55% MASCHI) FENOTIPO DEFICIENZA MENTALE, FACIES CARATTERISTICA, MALFORMAZIONI SCHELETRICHE, MALFORMAZIONI CARDIOVASCOLARI, DERMATOGLIFI ANORMALI, SOPRAVVIVENZA MEDIA INFERIORE ALLA NORMA
LO STUDIO DEI DERMATOGLIFI E’ IMPORTANTE IN PATOLOGIA NEONATALE E IN PEDIATRIA I DERMATOGLIFI DI INDIVIDUI DOWN HANNO CARATTERISTICHE DIAGNOSTICHE
CIRCA IL 70% DEI CONCEPIMENTI CON +21 NON RAGGIUNGE LA NASCITA • 92% TRISOMIA LIBERA PER NON DISGIUNZIONE PRIMARIA DURANTE LA GAMETOGENESI • 94% NON DISGIUNZIONE ALLA MEIOSI I MATERNA (TERMINALIZZAZIONE DEI CHIASMI, RIDOTTA FREQUENZA DI CROSSING OVER) • 5% TRISOMIA SECONDARIA A TRASLOCAZIONE ROBERTSONIANA • 46, XX o XY, D-, t(14q 21q)+ • 46, XX o XY, D-, t(Dq 21q)+ • 46, XX o XY, G-, t(Gq 21q)+
RELAZIONE TRA INCIDENZA DELLA SINDROME DI DOWN ED ETA' MATERNA FREQUENZA DI BAMBINI CON TRISOMIA 21: • NELLA POPOLAZIONE GENERALE 1/780 • IN DONNE AL DI SOTTO DEI 20 ANNI 1/1500 • IN DONNE OLTRE I 49 ANNI 1/8
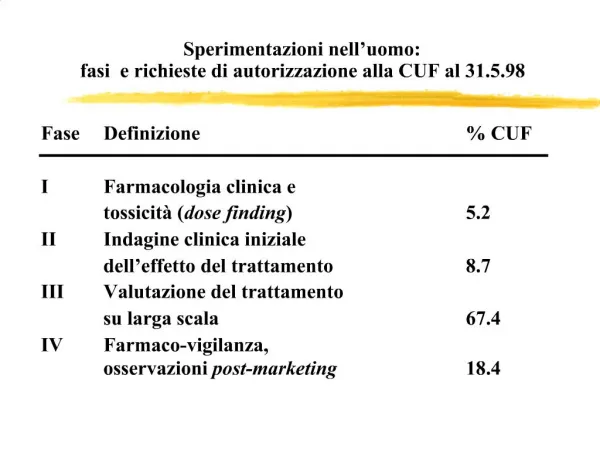
DIAGNOSI PRENATALE DI PATOLOGIE CROMOSOMICHE AMNIOCENTESI INTORNO ALLA 20A SETTIMANA CELLULE DEL LIQUIDO AMNIOTICO OSSIA CELLULE DI SFALDAMENTO FETALI VILLOCENTESI ENRO LA 12A SETTIMANA CELLULE DEL TROFOBLASTO TESSUTO FETALE PER L’IMPIANTO IN UTERO
TRASLOCAZIONE BILANCIATA 14;21 MEIOSI E GAMETI
CARIOTIPO DI UN PORTATORE SANO DI TRASLOCAZIONE BILANCIATA 14;21
CARIOTIPO DI UN PAZIENTE CON TRISOMIA SECONDARIA DERIVATA DA TRASLOCAZIONE BILANCIATA 14;21
DIAGNOSI DI TRISOMIA 21 MEDIANTE IBRIDAZIONE IN SITU CON SONDE DI DNA SU CROMOSOMI MITOTICI
IDENTIFICAZIONE DELLA TRISOMIA 21 SU NUCLEI INTERFASICI CON IBRIDAZIONE IN SITU
SINDROME DI EDWARDS47, XX XY, 18+ • FREQUENZA NEI NATI VIVI 1/3000 (80% FEMMINE) • CIRCA IL 95% DEI CONCEPIMENTI CON +18 NON RAGGIUNGE LA NASCITA • 90% TRISOMIA LIBERA PER NON DISGIUNZIONE PRIMARIA DURANTE LA GAMETOGENESI • 94% NON DISGIUNZIONE ALLA MEIOSI I MATERNA • LE TRISOMIE DI ORIGINE PATERNA SONO POST-ZIGOTICHE • <10% TRISOMIA A MOSAICO, DOPPPIA TRISOMIA, TRASLOCAZIONE FAMILIARE • IL FENOTIPO TIPICO E' DATO DALLA DUPLICAZIONE DI 18p E DELLA REGIONE PROSSIMALE DI 18q • SONO FREQUENTI ISOCROMOSOMI 18p + • SONO STATE DESCRITTE ANCHE SINDROMI 18p-, 18q-, 18r (RARISSIME)
SINDROME DI EDWARDS: FENOTIPO • GRAVE RITARDO DI SVILUPPO • IPOTONIA, SEGUITA DA IPERTONIA, APNEA, CONVULSIONI • MICROCEFALIA, FRONTE PROMINENTE • FACIES CARATTERISTICA: IPERTELORISMO, EPICANTO, MICROFTALMIA, ORECCHIE MALFORMATE AD IMPIANTO BASSSO • MALFORMAZIONI SCHELETRICHE: DITA CON ATTACCATURA ANORMALE, PIEDE CON PIANTA RICURVA, STERNO CORTO • MALFORMAZIONI CARDIOVASCOLARI • MALFORMAZIONI RENALI • GRAVE COMPROMISSIONE NEUROLOGICA • SOPRAVVIVENZA MEDIA 2-3 MESI (90% MUOIONO ENTRO IL PRIMO ANNO DI VITA)
SINDROME DI PATAU47, XX XY, 13+ • FREQUENZA NEI NATI VIVI (1/6000) • CIRCA IL 97% DEI CONCEPIMENTI CON +13 NON RAGGIUNGE LA NASCITA • 80% TRISOMIA LIBERA PER NON DISGIUNZIONE PRIMARIA DURANTE LA GAMETOGENESI • NELLA MAGGIOR PARTE DEI CASI NON DISGIUNZIONE ALLA MEIOSI I MATERNA • 20% TRISOMIA SECONDARIA A TRASLOCAZIONE ROBERTSONIANA: • 46, XX o XY, D-, t(13q 14q)+ • 46, XX o XY, D-, t(13q Dq)+ • 46, XX o XY, D-, t(13q Gq)+ • RARE TRISOMIE A MOSAICO, TRISOMIE PARZIALI
SINDROME DI PATAU: FENOTIPO • GRAVE COMPROMISSIONE NEUROLOGICA • GRAVE RITARDO DI SVILUPPO, RITARDO PSICOMOTORIO, IPOTONIA, CONVULSIONI • MICROENCEFALIA, LESIONI DELLO SCALPO • FACIES CARATTERISTICA: IPERTELORISMO, ANOFTALMIA O MICROFTALMIA, LABBRO LEPORINO, PALATOSCHISI, MICRORETROGNAZIA • MALFORMAZIONI SCHELETRICHE: POLIDATTILIA, PIEDE EQUINO MALFORMAZIONI CARDIOVASCOLARI, E RENALI • SOPRAVVIVENZA MEDIA < 5 MESI
SINDROME DA TRIPLOIDIA • INCIDENZA: 1/2500 NATI VIVI (MOSAICI) • LA TRIPLOIDIA E’ UNA CAUSA FERQUENTE DI ABORTO NEL 1°/2° TRIMESTRTE (20%) • INDIVIDUI TRIPLOIDI MUOIONI ENTRO I PRIMISSIMI GIORNI DI VITA • CARIOTIPO: • 69, XXY (60%) • 69, XXX (37%) • 69, XYY (<3%)
SINDROME DA TRIPLOIDIA:FENOTIPO • CRANIO A “GUSCIO D’UOVO ROTTO” • FACIES CARATTERISTICA: • NASO ADUNCO • MENTO PICCOLO • ORECCHIE MALFORMATE AD IMPIANTO BASSO • CAPELLI LANOSI
SINDROMI DA DELEZIONE CLASSICHE • DELEZIONE 5p (SINDROME CRI-DU-CHAT, DEL PIANTO DEL GATTO) • DELEZIONE 4p (SINDROME DI WOLF-HIRSCHORN, WHS) • SINDROMI DA MICRODELEZIONE • SINDROME DI WILLIAMS • SINDROME DI MILLER-DIEKER
SINDROME DEL PIANTO DEL GATTOCRI-DU-CHAT46, XX XY, 5p- • FREQUENZA NEI NATI VIVI (1/50.000) • CIRCA 80% DELEZIONE DE NOVO DI ORIGINE PATERNA • CIRCA 10% MALSEGREGAZIONE DI TRASLOCAZIONI PARENTALI
CRI-DU-CHAT: FENOTIPO • PIANTO FLEBILE SIMILE AL MIAGOLIO DI UN GATTO (LARINGE IPOPLASICA) • GRAVE RITARDO MENTALE E PSICOMOTORIO • MICROENCEFALIA • FACCIA A LUNA PIENA, IPERTELORISMO OCULARE, EPICANTO, MICROGNAZIA, MALFORMAZIONI SCHELETRICHE • SOPRAVVIVENZA MEDIA SIGNIFICATIVAMENTE RIDOTTA
SINDROME DI WOLF-HIRSCHORN (WHS) 46, XX XY, 4p- • RARISSIMA, INCIDENZA NON STIMABILE • DELEZIONE DE NOVO 90% DEI CASI • 10% DEI CASI SECONDARIA A TRASLOCAZIONE PARENTALE • CASI DI MOSAICISMO
SINDROME DI WOLF-HIRSCHORN:FENOTIPO • GRAVE RITARDO PSICOMOTORIO • MICROENCEFALIA • MICROFTALMIA • DIFETTI DELLO SCALPO • FACCIA AD “ELMO DI GUERRIERO GRECO” • ANOMALIE CARDIACHE, GENITO-URINARIE, SCHELETRICHE
SINDROME DI WILLIAMS46, XX XY, del (7) (q11.2) • FACIES DA ELFO: • GUANCE GONFIE • LABBRA CARNOSE • PHILTRUM ALLUNGATO E MOLLE • STENOSI DELL’AORTA • IPERCALCEMIA • IPERCALCIURIA • ANOMALIE MUSCOLO-SCHELETRICHE
SINDROME DI WILLIAMS: FENOTIPO
SINDROME DI MILLER-DIEKER45, XX XY, del(17) (p13.1-13.3) • GRAVE RITARDO MENTALE • LISSENCEFALIA (CERVELLO MOLLE) • CARATTERISTICHE DISMORFICHE