Download

1 / 12
200 likes | 883 Vues
Hydroformylation and oxidation of olefins. Textbook H: Chapter 16.6, 17.1 – 17.3 Textbook A: Chapter 16.1 – 16.2, 18.1 – 18.2. Outline. Hydroformylation Thermodynamics Mechanism Phosphine-modified catalysts Wacker reaction Monsanto process

E N D
Hydroformylation and oxidationof olefins Textbook H: Chapter 16.6, 17.1 – 17.3 Textbook A: Chapter 16.1 – 16.2, 18.1 – 18.2
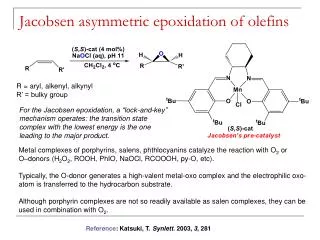
Outline • Hydroformylation • Thermodynamics • Mechanism • Phosphine-modified catalysts • Wacker reaction • Monsanto process • Katsuki-Sharpless asymmetric epoxidation of allylic alcohols • Jacobsen asymmetric epoxidation of olefins • Sharpless asymmetric dihydroxylation of olefins
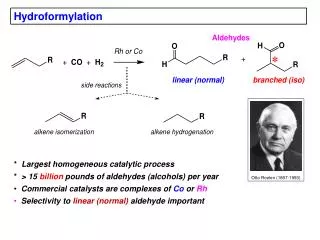
Hydroformylation: Thermodynamics Thermodynamics at standard conditions: At higher temperatures the entropy loss becomes more important: DG will be less negative. The TD favored product is the alkane. The obtained product is the aldehyde because of kinetic control.
Mechanism of the Co-catalyzed hydroformylation b-hydride elimination is suppressed due to the high CO pressure. Preferential formation of the linear product is due to steric interactions. All steps are reversible, except product formation, which is often rate-determining. Hydrogenolysis is the rate-determining step for simple terminal olefins. Trapping of the alkyl by CO determines the regioselectivity.
Other mechanistic details Last step Experimental evidence has been reported for both mechanisms under certain conditions. Heterometallic bimolecular formation of aldehyde has been observed. Direct reaction with H2 was slower than the manganese hydride reaction. Higher alkenes
Phosphine-modified catalysts • Alkylphosphines are strong electron donors and thus dissociation of CO is retarded, leading to more stable, but also slower catalysts. • A very effective ligand for Co is a phobane derivative: • The reaction is 100 times slower; • The selectivity to linear products increases; • The catalyst is more stable; • The catalyst acquires activity for hydrogenation • Order of activity (195 °C, 36 bar) • Ph2EtP > PhBu2P > Bu3P > Et3P > PhEt2P > Cy3P • Linear : branched ratio: • Bu3P > Et3P = PhEt2P = Cy3P = PhBu2P > Ph2EtP • For Rh, PPh3 works very well. HRh(CO)L3 complexes are 100 to 1000 times more active than Co complexes and they operate under milder conditions (15 – 25 atm and 80 °C – 120 °C).
Acetic acid and acetyl chemicals • Principal application of acetyl chemicals: solvents and vinyl acetate monomer for polymerization. • Early production routes (ca 1850s): fermentation or wood distillation (still used in the USA in the mid-1960s). • First synthetic route: Hg2+ catalyzed H2O addition to HC≡CH to form CH3CHO.
Vinyl acetate synthesis • Overall yields: 90 – 95% based on both ethylene and acetic acid. • Homogeneous process was abandoned due to corrosion associated with the presence of acetic acid. • Heterogeneous process uses PdCl2 / CuCl2 / C or PdCl2 / alumina.
Monsanto carbonylation of methanol The rate-determining step is the OA of MeI to Rh(I). Other metals are active; Ir: the CATIVA process; Ni, Pd. In the CATIVA process Me migration becomes rate-determining (addition of metal salts to abstract I-).
Katsuki-Sharpless asymmetric epoxidation of allylic alcohols Other tartrates are readily available. Small groups (COOEt) in b-position at considerable distance from the reactive center can induce high ee (> 98%): dimeric structure. Allylic tertiary alcohols are not successfully epoxidized. Substitution patterns: Reference: Pfenninger, A.Synthesis 1986, 2, 89
Mechanistic considerations Dimeric Ti complex before coordination of substrate The kinetics of the reaction are in accordance with replacement of two i-PrO anions by a t-butylperoxyanion and an allyloxy anion. This intermediate has a very low concentration and has not been observed directly. • The coordinated distal peroxo-O is transferred to the olefin. • The proximal peroxo-O interacts strongly with Ti in the TS. • The olefin pi* orbital must be in position to overlap with one of the lone pairs of the peroxo-O • that is being delivered. The attack is centered along the axis of the O-O sigma-bond being • broken. • The Ti alkoxide systems have several unique properties: • - Exchange of the monodentate alkoxide ligands is rapid in solution. • Ti(IV) has a somewhat flexible coordination sphere. • Ti(IV) alkoxides are weak Lewis acids: they activate to a certain extent a coordinated alkylperoxo ligand towards nucleophilic attack by the double bond of the allylic alcohol.