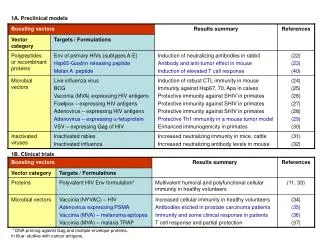
PRECLINICAL RESEARCH
PRECLINICAL RESEARCH. CLINICAL RESSEARCH PRODUCT DEVELOPMENT. PRECLINICAL RESEARCH. Phase I Phase II Phase III Phase IV. Chem-istry. Pharma-cology Toxi-cology. NDA**. IND*. GLP=Good Laboratory Practice Criteria for preclinical laboratory work. GCP=Good Clinical Practice


PRECLINICAL RESEARCH
E N D
Presentation Transcript
PRECLINICAL RESEARCH CLINICAL RESSEARCH PRODUCT DEVELOPMENT PRECLINICAL RESEARCH Phase I Phase II Phase III Phase IV Chem-istry Pharma-cologyToxi-cology NDA** IND* GLP=Good Laboratory Practice Criteria for preclinical laboratory work GCP=Good Clinical Practice Criteria for clinical trials GMP=Good Manufacturing Practice Criteria for pharmaceutical manufacture
NEW DRUG DEVELOPMENT A p p r o v a l Process Development Formulation/Stability PostFillingActivity Drug Delivery Drug Screening Pre-Clin.Workshop FileIND FileNDA Clinical Trial Phase I Phase II Phase III Metabolism Patient Process Toxicology PostSubmissionActivity Registration Workup Discovery Screening Pre-Clinical Development Ten Plus Years
廣義: Or • is defined as a prospective study comparing the effect and value of intervention(s) against a control in human subjects. (Fundamentals of Clinical Trials, Second edition, PSGPUBLISHING COMPANY, INC.,p2, 1985.)
臨 床 試 驗 規 範 倫理 利益 人體 品質 責任 分工
ICH: International Conference on Harmonization of Technical Requirements for the Registration of Pharmaceutics for Human Use.
Good Clinical Practice A standard for the design, conduct, performance, monitoring, auditing, recording, analysis and reporting of clinical trials that provides assurance that the data and reported results are credible and accurate and that the rights, integrity, and confidentiality of trial subjects are protected. ICH GCP Glossary 1.24
藥品優良臨床試驗規範 Good Clinical Practice 衛生署 85.11.20 公告 GMP, GLP, ISO……...
壹、總則 貳、醫療機構 參、受試驗者之保護措施 肆、試驗主持人 伍、試驗委託者 陸、監測者 柒、紀錄與報告 捌、統計分計 玖、研究用藥品之管理 拾、品質保證 藥品優良臨床試驗規範
總則-名詞定義(I) • 藥品優良臨床試驗規範 <Good Clinical Practice, GCP> • 臨床試驗 <Clinical Trial> • 研究用藥品 <Study Product> • 人體試驗委員會 <Ethics Committee > • 試驗計畫書 <Protocol> • 試驗主持人 <Investigator> • 總主持人 <Principle Investigator> • 藥品不良反應 <Adverse Drug Reaction> • 受試驗者 <Trial Subject> • 試驗委託者 <Sponsor>
總則-名詞定義(II) • 受試同意書 <Informed Consent> • 多機構合作臨床試驗 <Multi-Centre Trials> • 主持人手冊 <Investigator’s Brochure> • 監測者 <Monitor> • 不良事件 <Adverse Event, AE> • 資料驗證 <Verification or validation of data> • 試驗稽核 <Audit of a Trial> • 查核 <Inspection> • 隱私權 <Confidentiality>
總則-名詞定義(III) • 個案報告表 <Case Report Form> • 合約 <Contract> • 受託研究機構 <Contract Research Organization, CRO> • 藥品優良製造規範 <Good Manufacturing Practice, GMP > • 標準作業程序 <Standard Operating Procedures, SOP> • 總結報告 <Final Report> • 原始資料 <Raw Data>
醫療機構 • 具備良好的設備和編制的醫療機構進行臨床觀察、檢驗及緊急時的必要治療。 • 設立人體試驗委員會以審查臨床試驗之進行。 • 對臨床試驗的執行(修正)給予意見,並批准臨床試驗計畫書(的修正),報備中央衛生主管機關核准後,始予進行。 • 在得知有嚴重不良反應後,應監督試驗主持人進行必要的措施。
臨床試驗申請步驟 具備申請資料 人體試驗委員會審查 衛生署審查 同意後始得進行
人體試驗委員會 • 組成:最少五人,包括男性、女性及非專業人士 • 運作:定期開會審查,書面紀錄審查意見、會議情形 及同意試驗進行 • 保護受試者權益:審查主持人的能力是否恰當、計畫 設計是否週全可行、因參加試驗導 致傷害時,治療、救濟、保險、賠 償是否完善
ICH GCP Requirements for the composition of the Ethics Committee(IRB) “A reasonable number of members who collectively have the qualifications and experience to review and evaluate the science, medical aspects and ethic of the proposed trial.” • At least 5 members • At least one member whose primary interest is non-scientific (lay member) • At least one member who is independent of the trial site. Only members who are independent of the investigator can vote or provide an opinion.
The Principles of ICH GCP • According to the Declaration of Helsinki • Risk benefit evaluation before start • The interest of patient before that of science cf. guideline, regulation, law
受試驗者之保護措施 受試者同意書 • 須符合赫爾辛基宣言,保護受試驗者的個人權益。 • 除給予受試驗或法定代理人書面資料外,並包括口頭說明與雙向溝通,使其了解整個試驗的狀況,並有充裕的時間考慮後,再決定簽署受試同意書。
受試者同意書 內容: 1.試驗目的及方法 2.可能產生之副作用及危險 3.預期試驗效果 4.其他可能之治療方式及說明 5.受試驗者無須提出任何理由,即得隨時撤回同意,退出試驗
受試者同意書 • 由受試驗者或其法定代理人的簽署,並載明日期始生效力。 • 試驗委託者應為參加臨床試驗的病患及健康自願者加入適當的保險,以保障可能遭受的任何傷害。
Informed Consent • The FDA has suspended all seven gene therapy clinical trial at the University of Pennsylvania due to possible serious protocol and informed consent violations.
Sponsor Investigator(s) IRB Patients General Interactions
試驗主持人 資格 • 該藥品類屬科別之專科醫師。 • 醫療專業領域裡具備有良好的知試和經驗。 • 熟悉臨床試驗的研究方法。 • 明瞭試驗委託者提供的資料,文獻和資訊。 • 明瞭並遵法規和倫理的要求。
責任(I) • 完全熟悉主持人手冊所記載研究用藥品的性質 • 保證具有充份的時間、適當的工作人員和設施完成試驗 • 確保受試驗者募集的速度 • 提供最新的學經歷資料 • 與試驗委託者共同簽署試驗計畫書 • 書面保證遵守試驗計畫書及藥品優良臨床試驗規範 • 接受監測者和管制程序的監督,並遵守雙方有關論文發表之協議
責任(II) • 向人體試驗委員會提出試驗計畫之申請 • 提供試驗工作人員有關臨床試驗及受試驗者處理的資訊 • 進入臨床試驗前,向受試驗者或法定代理人說明,並取得受試同意書 • 建立研究用藥品標準作業程序 由藥師點收、運送過程、貯存、僅供試驗用、剩餘歸還 • 謹慎處理編碼之過程並加以記錄 • 保密: 研究資料、受試驗者的隱私權、試驗委託者提供之資料
責任(III) • 正確收集資料、紀錄 、保存,供做成報告並供驗證、 稽核或查核。 • 與分析者共同簽署之試驗資料、 結果和分析報告,由醫療機構送至試驗委託者 • 嚴重不良事件發生時,應立即通知: • 試驗委託者 • 人體試驗委員會 • 醫療機構首長 • 中央衛生主管機關 並對受試驗者採取適當的保護措施
Disqualification of a clinical investigator(FDA 21 CFR 312.70) 1) repeated & deliberate submission of false data 2) failure to obtain IRB approval & comply with IRB regulation 3) failure to obtain informed consent 4) failure to sign investigator statement 1572/1572s 5) failure to follow protocol - research plan 6) failure to administer drug/ or under PI supervision 7) inadequate recordkeeping requirements 1. Drug dispensing 2. Adequate case histories 3. Record retention 8) refuse to show inspection of records
Tips for an effective clinical investigator • Familiar with protocol to fit usual practice • Patient pool and data base • recruitment target and solve problems in time • Full time study nurse and coordinator • Build up an experienced and efficient team • IRB relationships and GCP guidelines • Update C.V. • Enough time, money and regular meetings • Source data verification • Complete CRF correctly and keep all documents • Avoid major trial deficiencies • Never invent data
試驗委託者 - 責任 (I) • 建立詳細的書面標準作業程序(SOP) • 選擇適當主持人 - 試驗場所、設施、資格、能力 • 同意試驗主持人相關責任之分工 - 資料處理、揭露代碼、統計處理、試驗報告的準備,並載於試驗計畫書,共同簽署
試驗委託者 - 責任 (II) • 提供主持人手冊 • 提供最新試驗藥品資料 • 提供完整且正確代碼編號的研究用藥品 • 指派合適的個人或委員會,執行和監測(monitor)試驗 • 建立品質保證的體系,包括獨稽核(audit),以確保試驗的進行和資料的產生、抄錄和報告均符合試驗計畫書
試驗委託者 - 責任 (III) • 如發生和試驗相關的傷害或死亡的事件,應和醫療機構及試驗主持人提供受試驗者適當的治療或救濟及賠償,並提供該試驗的不良事件報告表及通報 • 決定或必須提前結束試驗時,應將此決定和其理由告知試驗主持人、人體試驗委員會和中央衛生主管機關 • 確定所提出的總結報告符合規定
監測者 - 責任 (I) • 由試驗委託者指派,協助確認試驗進行符合計畫書及GCP • 為試驗委託者及試驗主持人之間的主要溝通連繫者 • 根據標準作業程序執行,訪視試驗主持人-試驗進行,確認資料紀錄與報告是否完整,受試同意書簽署
監測者 - 責任 (II) • 以原始資料核對個案報告表,並告知試驗主持人 • 與試驗主持人每次之會面、電話及信件等討論後,均應有一份書面的監測紀錄 • 協助試驗主持人進行必須的報告
Correcting data in a CRF 5 4 DHR 12/12/97 • Cross out wrong entry with a single line • Do not use correcting fluids • Write correct entry alongside • Initial and date change
Some golden rules for CRF completion Please tick box when appropriate item has been undertaken • Check each page of the CRF when completed; initial and date all errors or alterations (cheque-book style). • Read and follow all instruction carefully. • Complete All Sections ;try to avoid missing data. • If data is to be entered retrospectively, remember to do it. • If you need to alter data, strike through the incorrect value with a single line(leaving the original data legible), enter the new data alongside, initial and date and give a reason for the change.. • Keep the original copies of all data transcribed into the record form for later verification by the monitor. • Never use correcting fluids. • Sing the CRF as requested (each page or at the end) to confirm it is a true record. • Try to write legibly, using a black pen to facilitate photocopying.
紀錄與報告-(I) • 資料檔案之保管 • 至少七年,由試驗委託者、主持人及相關場所 • 供主管機關查核(inspection)並得影印或以其他方法複製該紀錄或其副本 • 個案報告表 • 試驗中任何觀察與發現均正確且完整的記錄,紀錄者並應簽署 • 更正時不得消除原始登錄,簽署日期及原因
紀錄與報告-(II) • 電腦資料-使用權限,登錄,更正 • 受試驗者身份 • 試驗主持人應持有一份詳盡、保密及隨時可確認受試驗者身份的紀錄。 • 試驗委託者必須能藉由清楚的代碼確認每個受試驗者的試驗數據。
統計分析-(I) • 臨床試驗宜有生物統計學專家參與 • 隨機分配及盲性試驗 • 隨機分配受試驗者的過程必須加以紀錄,每個受試驗者的密封代碼應由試驗主持人及試驗委託者雙方同時保存。 • 當進行盲性試驗時,應清楚註明揭露代碼的時機。代碼揭露時間及其理由須載明於個案報告表上。
統計分析-(II) • 統計分析時應注意事項: • 統計分析方法及主要療效指標必須在試驗計畫書中指明。期間分析的可行性及時機亦必須於試驗計畫書中指明。受試驗者數目之預估及說明其統計檢定力,亦須詳載於試驗計畫書。 • 統計分析結果應著重於臨床上重要性的說明。 • 統計過程中發現疏漏、未使用及多餘的資料,均須加以說明。
關係 衛生主管機關 試驗委託者 試驗主持人 人體試驗委員會 受試者
研究用藥品的管理 • 試驗委託者負責供應 • 依規定標示或說明製造日期、使用期限、用量紀錄、運送、供應、貯藏方式。 • 管理紀錄中應包括數量、運送情形、收據、配置、收回、餘藥的銷毀等資訊 • 不得將藥品供應給非受試驗者 • 標明 “臨床試驗專用” 。 • 由試驗委託者保存至新藥核准上市後二年,其未申領藥品許可證者,亦應保存至臨床試驗結束後二年。
品質保證 • 所有觀察結果與發現,資料的可靠性均得被再確認 • 確保所有資料數據之可靠及被正確處理 • 試驗委託者監測 • 由與試驗計畫無關之人員執行稽核 • 中央衛生主管機關查核
國內臨床試驗 • 衛生主管機關 行政院衛生署-醫政處/藥政處 • 相關法規/罰責 醫療法及其施行細則-人體試驗範圍、執行 藥事法-試驗用藥物、新藥規定 藥物樣品贈品管理辦法-申請進口試驗用藥物 公告-解釋、規範
臨床試驗基準 • 衛生署已公告: • 1. 藥品臨床試驗一般基準(1999.03) • 2. 核醫放射性藥品臨床試驗基準(1999.05) • 3. 心血管治療藥品臨床試驗基準(1999.10) • 4. 感染症治療藥品臨床試驗基準(1999.10) • 5. 癌症治療藥品臨床試驗基準(1999.12) • 6. 內分泌及新陳代謝治療藥品臨床試驗基準(1999.12) • 7. 植物抽取新藥臨床試驗基準(2000.03)