CLL, HCL, CML
CLL, HCL, CML. Mirzania.MD pathophysiology. Epidemiology and clinical manifestations of chronic lymphocytic leukemia. Chronic lymphocytic leukemia (CLL) is one of the chronic lymphoproliferative disorders (lymphoid neoplasms).


CLL, HCL, CML
E N D
Presentation Transcript
CLL, HCL, CML Mirzania.MD pathophysiology
Epidemiology and clinical manifestations of chronic lymphocytic leukemia
Chronic lymphocytic leukemia (CLL) is one of the chronic lymphoproliferative disorders (lymphoid neoplasms). • It is characterized by a progressive accumulation of functionally incompetent lymphocytes, which are monoclonal in origin.
EPIDEMIOLOGY • CLL is the most common leukemia in Western countries • The disorder is more common in men with a male to female ratio of approximately 1.7:1. • CLL is considered to be mainly a disease of the elderly, with a median age at diagnosis of 70 years. • Genetic rather than environmental factors are the most likely explanation for these differences. • higher incidence among Caucasians as compared with African Americans or Asian Pacific Islanders
CLINICAL PRESENTATION • Symptoms • painless swelling of lymph nodes, often in the cervical area, which spontaneously wax and wane, but do not altogether disappear. • Approximately 25 percent of patients feel entirely well with no symptoms when a routine blood count reveals an absolute lymphocytosis, leading to a diagnosis of CLL. • Five to 10 percent of patients present with the typical "B" symptoms .
Symptoms • an acquired immunodeficiency disorder, manifested by infections, • autoimmune complications such as hemolytic anemia, thrombocytopenia or pure red cell aplasia, or exaggerated reactions to insect stings or bites (especially mosquito).
Signs • Lymphadenopathy — The most common abnormal finding present in 50 to 90 percent of patients among various series. • enlargement may be generalized or localized • The most commonly affected sites are cervical, supraclavicular, and axillary. • enlarged nodes in CLL are firm, rounded, discrete, nontender, and freely mobile upon palpation.
Signs • Splenomegaly — The spleen is the second most frequently enlarged lymphoid organ, being palpably enlarged in 25 to 55 percent of cases. • usually painless, and nontender to palpation, with a sharp edge and a smooth firm surface. • Hepatomegaly — Enlargement of the liver may be noted at the time of initial diagnosis in 15 to 25 percent of cases.
Signs • Skin — Infiltration in the skin (leukemia cutis) is the most commonly involved non-lymphoid organ. These lesions most commonly involve the face and can manifest as macules, papules, plaques, nodules, ulcers, or blisters • Nonspecific secondary cutaneous lesions may be due to infection, bleeding, vasculitis, or paraneoplasticpemphigus. • Membranoproliferativeglomerulonephritis — Membranoproliferativeglomerulonephritis (MPGN) has occasionally been described in CLL, and appears to be a paraneoplastic phenomenon mediated by deposition and possibly processing of cryoprecipitating or noncryoprecipitating M-components .
Signs • Other organ involvement — Virtually any lymphoid tissue may be enlarged at diagnosis, including • Waldeyer's ring in the pharynx. • In contrast to other lymphomas, gastrointestinal mucosal involvement is rarely seen in CLL. • Similarly, meningeal leukemia is unusual at the time of initial presentation.
LABORATORY ABNORMALITIES • Lymphocytosis:the absolute blood lymphocyte threshold for diagnosing CLL has been placed at >5000/microL [5 x 10(9)/L] B lymphocytes , a significant proportion of patients present with counts as high as 100,000/microL [100 x 10(9)/L]. • Cytopenias — Neutropenia, anemia and thrombocytopenia may be observed at the time of initial diagnosis, and are usually not severe. These can be related to autoimmune hemolytic anemia, pure red cell aplasia, autoimmune thrombocytopenia, or agranulocytosis.
LABORATORY ABNORMALITIES • Immunoglobulin abnormalities — Hypogammaglobulinemia is present in approximately 8 percent of patients at the time of initial diagnosis and may develop in up to two-thirds of patients later in the course of the disease. Usually all three immunoglobulin classes (IgG, IgA, and IgM) are decreased. • Polyclonal increases in gamma globulins can be seen in up to 15 percent of patients, while a monoclonal protein, usually of the same class as the surface membrane immunoglobulin, is present in up to 5 percent of patients.
LABORATORY ABNORMALITIES • Other abnormal findings • elevated levels of serum lactate dehydrogenase (LDH) and beta-2 microglobulin were found in 60 percent of patients in one series. • Elevations of uric acid, hepatic enzymes (ALT or AST) and, rarely, calcium may also be observed.
immunophenotype • Expression of one or more B-cell associated antigens as demonstrated by CD19, CD20, CD21, CD23, and CD24. • Expression of CD5, a T-cell associated antigen
Staging and prognosis of chronic lymphocytic leukemia • NATURAL HISTORY • prolonged (ie, 10 to 20 years) clinical course(1/3). • Some patients die rapidly, within two to three years from diagnosis. • Other patients live for 5 to 10 years with an initial course that is relatively benign followed by a terminal phase lasting one to two years. During this terminal phase there is considerable morbidity, both from the disease itself and from complications of therapy. • during the terminal phase the performance status is poor, with recurring need for hospitalization. The most frequent causes of death are severe systemic infection (especially pneumonia and septicemia), bleeding, and inanition with cachexia.
CLINICAL STAGING AND PROGNOSIS the natural history of CLL is extremely variable, with survival times from initial diagnosis that range from 2 to 20 years, and a median survival of approximately 10 years.
Rai stage • Stage 0 — 150 months • Stage I — 101 months • Stage II — 71 months • Stages III and IV — 19 months
HISTOLOGIC TRANSFORMATION • Prolymphocytic leukemia — 10 percent • Aggressive or highly aggressive lymphoma (Richter's transformation) — 3 percent • Hodgkin lymphoma — 0.5 percent • Multiple myeloma — 0.1 percent
Prolymphocytoid transformation • In approximately 10 percent of patients with CLL, the terminal event is a morphological transformation of blood lymphocytes • from the typical small, mature-appearing cell • to somewhat larger cells with distinct nucleoli and a less dense nuclear chromatin. This event, called prolymphocytoid transformation, occurs slowly over years and is associated with refractoriness to the usual chemotherapeutic agents.
Richter's syndrome or Richter's transformation • In 1 to 10 percent of patients with CLL, transformation to a highly aggressive non-Hodgkin lymphoma supervenes. • This transformation is heralded by sudden clinical deterioration, characterized by increasing lymphadenopathy, splenomegaly, worsening "B" symptoms, a rapidly progressive downhill clinical course, and a median survival of 5 to 8 months.
transformation • Acute leukemic transformation — Acute leukemia is a rare terminal event in CLL and, when it does occur, is usually one of the myeloid variants (ie, acute myeloid leukemia, AML). • Prior therapy with alkylating agents has not been clearly implicated in causation of terminal leukemia. • Second malignancies — Patients with CLL are at a higher risk than the general population to develop another cancer, usually of the lung, skin, and bone
INDICATIONS FOR TREATMENT • With the possible exception of allogeneic hematopoietic cell transplantation (HCT), CLL cannot be cured by current treatment options. • Prospective, randomized trials evaluating immediate versus delayed treatment strategies have found no improvement in long-term survival with early treatment.
Therapy is indicated for patients with the following disease-related complications • Weakness, night sweats, weight loss, painful lymphadenopathy, or fever. • Symptomatic anemia and/or thrombocytopenia. • Autoimmune hemolytic anemia and/or thrombocytopenia poorly responsive to corticosteroid therapy. • Progressive disease, as demonstrated by increasing lymphocytosis with a lymphocyte doubling time less than six months, and/or • rapidly enlarging lymph nodes, spleen, and liver. • Repeated episodes of infection. Hypogammaglobulinemia without repeated episodes of infection is not a clear indication for therapy.
Clinical features and diagnosis of hairy cell leukemia • named HCL in the 1960s because of the prominent irregular cytoplasmic projections of the malignant cells. • The clinical features and diagnosis of HCL, which is now considered one of the indolent non-Hodgkin lymphomas.
INCIDENCE AND PATHOGENESIS • Hairy cell leukemia is an uncommon malignancy, representing about 2 percent of all leukemias. • The median age at onset is 52; • there is a strong male predominance of about four to one. • The incidence is about three times higher in Caucasians than in Blacks
The pathogenesis of HCL is unknown, although • exposure to ionizing radiation, • Epstein-Barr virus, • organic chemicals, • woodworking, and • farming
The cell expresses pan B cell surface antigens (CD19, CD20, and CD22) and an early plasma cell marker (PCA-1). It does not express CD21, a marker for the immature B cell. • The cell also expresses surface antigens that are not common on B cells, such as CD11c (monocytes and neutrophils), CD25 (activated T cells), and CD103 (intraepithelial T cells). • Clonalkaryotypic abnormalities are present in approximately two-thirds of patients. Abnormalities of chromosome 5 are present in approximately 40 percent
CLINICAL PRESENTATION • Approximately one-quarter present with abdominal fullness or discomfort due to splenomegaly, which may be massive. Spontaneous splenic rupture may occur, which constitutes a medical emergency. • Another one-quarter present with systemic complaints such as fatigue, weakness, and weight loss. Patients do not usually complain of fever or night sweats.
CLINICAL PRESENTATION • Another one-quarter present either with bruising and bleeding secondary to severe thrombocytopenia, or with recurrent infections, which may be life-threatening, secondary to granulocytopenia and monocytopenia. • The remaining one-quarter are generally asymptomatic and come to the clinician's attention because of an incidental finding of splenomegaly or cytopenias during evaluation for an unrelated cause. • Occasional patients have vasculitis, usually polyarteritis nodosa or cutaneous leukocytoclastic vasculitis, or other autoimmune manifestations
Physical examination • palpable splenomegaly in 80 to 90 percent of cases, with the splenic edge extending more than 8 cm below the left costal margin in 25 percent. • Hepatomegaly and lymphadenopathy are not major features of HCL, being present in about 20 and 10 percent of patients, respectively. • Rare physical findings include soft tissue infiltration, vasculitic skin rash, ascites, and pleural effusion.
Laboratory findings • Sixty to 80 percent of patients with HCL present with pancytopenia, with hematocrits in the range of 20 to 35 percent, a total white blood cell count usually below 4000/microL, and platelet counts in the range of 20,000 to 100,000/microL. • Abnormal liver function tests — 20 percent • Hypergammaglobulinemia — 20 percent • Leukocytosis (total WBC >10,000/microL) — 10 to 20 percent
DIAGNOSIS • The abnormal cell in HCL about 90 percent of patients. • Bone marrow examination . Because of fibrosis, the BM is often inaspirable, resulting in a "dry tap. • Reticulin — Staining of the BM trephine biopsy for reticulin almost always shows a moderate to marked increase in reticulin fibers. • Cytochemical findings — Historically, demonstration of tartrate-resistant acid phosphatase (TRAP) activity on peripheral blood films, marrow aspirate smears, or touch preparations of BM was routinely used to confirm the diagnosis of HCL. TRAP-positive cells are found in almost all cases of HCL at diagnosis
Hairy cells strongly express pan-B cell antigens including CD19, CD20, CD22, and CD25, and • usually lack expression of CD5, CD10, CD21, and CD23. • Hairy cells characteristically express CD11c, CD103, CD123, cyclin D1, and annexin A1.
INDICATIONS FOR TREATMENT • Many patients with HCL are asymptomatic and can be observed for months or years after the diagnosis is established before requiring treatment. • Significant cytopenias; typical peripheral blood counts that warrant treatment include an absolute neutrophil count <1000/microL with repeated infections, symptomatic anemia with a hemoglobin concentration <11.0 g/dL, or bleeding due to a platelet count <100,000/microL • Symptomatic splenomegaly (common) or symptomatic adenopathy (uncommon) • Constitutional symptoms (eg, fever, night sweats, fatigue, weight loss)
chronic myeloid leukemiaCML • A myeloproliferative neoplasm characterized by the dysregulated production and uncontrolled proliferation of mature and maturing granulocytes with fairly normal differentiation.
INTRODUCTION • CML is associated with the fusion of two genes: BCR (on chromosome 22) and ABL1 (on chromosome 9) resulting in the BCR-ABL1 fusion gene. • This abnormal fusion typically results from a reciprocal translocation between chromosomes 9 and 22, t(9;22)(q34;q11), that gives rise to an abnormal chromosome 22 called the Philadelphia (Ph) chromosome. • It is this derivative chromosome 22 which harbors the BCR-ABL1 fusion gene.
INTRODUCTION • The BCR-ABL1 fusion gene results the BCR-ABL1 fusion protein. • This protein product includes an enzymatic domain from the normal ABL1 with tyrosine kinase catalytic activity, but relative to ABL1, whose kinase activity is tightly regulated [1], • the kinase activity of BCR-ABL1 is elevated and constitutive [2] due to fusion with a portion of BCR. • It is this deregulated tyrosine kinase that is implicated in the pathogenesis of CML.
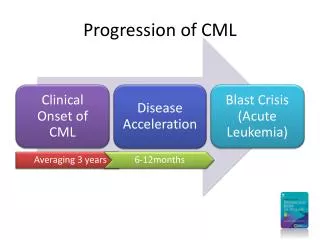
INTRODUCTION • The clinical hallmark of CML is the uncontrolled production of mature and maturing granulocytes, predominantly neutrophils, but also basophils and eosinophils. • In the absence of treatment, CML has a triphasic or biphasic clinical course as it progresses from a chronic phase to an accelerated phase and on to a terminal blast crisis. • Sometimes it goes from chronic phase directly to blast crisis, particularly when the blast phase is lymphoid.
EPIDEMIOLOGY • CML accounts for approximately 15 to 20 percent of leukemias in adults. • It has an annual incidence of 1 to 2 cases per 100,000, with a slight male predominance. • The median age at presentation is approximately 50 years for patients enrolled on clinical studies. • Exposure to ionizing radiation is the only known risk factor. • There is no known familial predisposition.
CLINICAL MANIFESTATIONS • CML has a triphasic or biphasic clinical course: • a chronic phase, which is present at the time of diagnosis in approximately 85 percent of patients; • an accelerated phase, in which neutrophil differentiation becomes progressively impaired and leukocyte counts are more difficult to control with treatment; and • blast crisis, a condition resembling acute leukemia in which myeloid or lymphoid blasts proliferate in an uncontrolled manner.
CLINICAL MANIFESTATIONS • Twenty to 50 percent of patients are asymptomatic, with the disease first being suspected from routine blood tests. • Among symptomatic patients, • fatigue (34 percent), • malaise (3 percent), • weight loss (20 percent), • excessive sweating (15 percent), • abdominal fullness (15 percent), and • bleeding episodes due to platelet dysfunction (21 percent) are common.
CLINICAL MANIFESTATIONS • Abdominal pain and left upper quadrant pain (sometimes referred to the left shoulder) and • early satiety, due to the enlarged spleen with or without • splenic infarction. • Tenderness over the lower sternum, due to an expanding bone marrow, is sometimes seen. • Acute gouty arthritis may also present at this time, due to overproduction of uric acid.
CLINICAL MANIFESTATIONS • splenomegaly (present in 48 and 76 percent in two series), • anemia (45 and 62 percent), • white blood cell count above 100,000/microL (52 and 72 percent), and • platelet count above 600,000 to 700,000/microL (15 and 34 percent). • Involvement of extramedullary tissues such as the lymph nodes, skin, and soft tissues is generally limited to patients with blast crisis.