HEME SYNTHESIS
E N D
Presentation Transcript
HEME SYNTHESIS & DISORDERS M.Prasad Naidu MSc Medical Biochemistry, Ph.D.Research Scholar
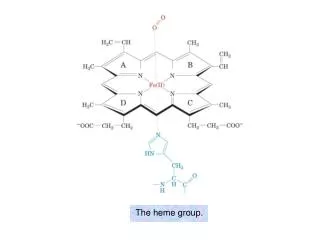
HEME SYNTHESIS • Heme is the most important porphyrin containing compound. • Heme is a derivative of the porphyrin. • Porphyrins are cyclic compounds formed by fusion of 4 pyrrole rings linked by methenyl (=CH-) bridges.
Metal ions can bind with nitrogen atoms of pyrrole rings to form complexes. • Since an atom of iron is present, heme is a ferroprotoporphyrin. • The pyrrole rings are named as l, ll, lll, lV and the bridges as alpha, beta, gamma and delta.
Naturally occurring porphyrins contain substituent groups replacing the 8 hydrogen atoms of the porphyrin nucleus. • When the substituent groups have a symmetrical arrangement (1, 3, 5, 7 and 2, 4, 6, 8) they are called the I series –type l porphyrins. • The lll series have an asymmetrical distribution of substituent groups (1, 3, 5, 8 and 2, 4, 6, 7)-type ll porphyrins.
Type lll is the most predominant in biological systems. • It is also called series 9, because fischer, the pioneer in porphyrin chemistry has placed it as the 9th in a series of 15 possible isomers. • Hans Fischer, the father of porphyrin chemistry, proposed a short hand model for presentation of porphyrin structures.
Hans Fischer synthesisedheme in laboratory in 1920(Nobel prize, 1930). • The usual substitutions are : a.propionyl (-CH2-CH2-COOH) group b. acetyl (-CH2-COOH) group c. methyl (-CH3) group d. vinyl (-CH=CH2) group
Biosynthesis of Heme • Heme can be synthesised by almost all the tissues in the body. • Heme is primarily synthesised in the liver and the erythrocyte-producing cells of bone marrow (erythroid cells). • Heme is synthesised in the normoblasts, but not in the matured ones.
The pathway is partly cytoplasmic and partly mitochondrial. Step 1: ALA synthesis • The synthesis starts with the condensation of succinyl CoA and glycine in the presence of pyridoxal phosphate to form delta amino levulinic acid (ALA). • The enzyme ALA synthase is located in the mitochondria and is the rate-limiting enzyme of the pathway.
Step 2: Formation of PBG • Next few reactions occur in the cytoplasm. • Two molecules of ALA are condensed to form porphobilinogen (PBG). • The condensation involves removal of 2 molecules of water and the enzyme is ALA dehydratase. • Porphobilinogen is a monopyrrole. • The enzyme contains zinc and is inhibited by Lead.
Step 3: Formation of UPG • Condensation of 4 molecules of the PBG, results in the formation of the first porphyrin of the pathway, namely uroporphyrinogen(UPG). • The pyrrole rings are joined together by methylene bridges, which are derived from alpha carbon of glycine.
When the fusion occurs, the lll series of isomers are predominantly formed; and only the lll series are further used. • This needs 2 enzymes which catalyse the reactions; PBG-deaminase (Uroporphyrinogen-l-synthase) and Uroporphyrinogen-lll-cosynthase. • During this deamination reation 4 molecules of ammonia are removed.
Step 4: synthesis of CPG • The UPG-lll is next convertedto coproporphyrinogen (CPG-lll) by decarboxylation. • Four molecules of CO2 are eliminated by uroporphyrinogen decarboxylase. • The acetate groups (CH2-COOH) are decarboxylated to methyl (CH3) groups.
Step 5: synthesis of PPG • Further metabolism takes place in the mitochondria. • CPG is oxidised to protoporphyrinogen (PPG-lll) by coproporphyrinogen oxidase. • Two propionic acid side chains are oxidatively decorboxylated to vinyl groups.
Step 6: Generation of PP • The Protoporphyrinogen-lll is oxidised by the enzyme protoporphyrin-lll (PP-lll) in the mitochondria. • The oxidation requires molecular oxygen. • The methylene bridges (-CH2) are oxidised to methenyl bridges (-CH=) and coloured porphyrins are formed. • Protoporphyrin-9 is thus formed.
Step 7: Generation of Heme • The last step in the formation of heme is the attachment of ferrous iron to the protoporphyrin. • The enzyme is heme synthase or ferrochelatase which is also located in mitochondria. • Iron atom is co-ordinately linked with 5 nitrogen atoms (4 nitrogen of pyrrole rings of protoporphyrin and 1st nitrogen atom of a histidine residue of globin).
The remaining valency of iron atom is satisfied with water or oxygen atom. • When the ferrous iron (Fe++) in heme gets oxidised to ferric (Fe+++) form, hematin is formed, which loses the property of carrying the oxygen. • Heme is red in colour, but hematin is dark brown.
Regulation of Heme synthesis • ALA synthase is regulated by repression mechanism. • Heme inhibits the synthesis of ALA synthase by acting as a co-repressor. • ALA synthase is also allosterically inhibited by hematin. • When there is excess of free heme, the Fe++ is oxidised to Fe+++(ferric), thus forming hematin.
The compartmentalisation of the enzymes in the synthesis of heme makes it easier for the regulation. • The rate-limiting enzyme is in the mitochondria. • The steps 1,5,6, and 7 are taking place inside mitochondria, while steps 2,3 and 4 are in cytoplasm.
Drugs like barbiturates induce heme synthesis. • Barbiturates require the heme containing cytochrome p450 for their metabolism. • Out of the total heme synthesised, two thirds are used for cytochrome p450 production. • The steps catalysed by ferrochelatase and ALA dehydratase are inhibited by lead.
INH (Isonicotinic acid hydrazide) that decreases the availability of pyridoxal phosphate may also affect heme synthesis. • High cellular concentration of glucose prevents induction of ALA synthase. • This is the basis of glucose to relieve the acute attack of porphyrias.
Shunt Bilirubin • When 15N or 14C labelled glycine is injected, this is incorporated into heme and into RBCs. • After 100-120 days, when RBCs are lysed, the radiolabelled Hb level is decreased, along with consequent rise in radioactive bilirubin. • However, about 15% of radioactive bilirubin is excreted within about 10 days. • This is called Shunt bilirubin.
This is the formation of bilirubin from heme in bone marrow, without being incorporated into Hb. • This is the result of ineffective erythropoiesis. • In porphyrias, especially in the erythropoietic varieties, the shunt biliribin will be increased.
Disorders of Heme synthesis • Porphyrias are group of inborn errors of metabolism associated with the biosynthesis of heme.(Greek ‘porphyria’ means purple). • These are characterised by increased production and production and excretion of porphyrins and/or their precursors (ALA + PBG). • Many of the porphyrias are inherited as autosomal dominant traits.
Porphyrias may be broadly grouped into 3 types: • Hepatic porphyrias b. Erythropoieticporphyrias c. porphyrias with both erythropoietic and hepatic abnormalities.
Acute intermittent porphyria This disorder occurs due to the deficiency of the enzyme uroporphyrinogen l synthase. Acute intermittent porphyria is characterised by increased excretion of porphobilinogen and δ-aminolevulinate. The urine gets darkened on exposure to air due to the conversion of porphobilinogen to porphobilin and porphyrin.
It is usually expressed after puberty in humans. Clinical features • The symptoms include abdominal pain, vomiting and cardiovascular abnormalities. • The neuropsychiatric disturbances observed in these patients are believed to be due to reduced activity of tryptophan pyrrolase (caused by depleted heme levels), resulting in the accumulation of tryptophan and 5-hydroxytryptamine.
These patients are not photosensitive since the enzyme defect occurs prior to the formation of uroporphyrinogen. • The symptoms are more severe after administration of drugs (e.g. barbiturates) that induce the synthesis of cytochrome P450. • This is due to the increased activity of ALA synthase causing accumulation of PBG and ALA.
Treatment: • Acute intermittent porphyria is treated by administration of hematin which inhibits the enzyme ALA synthase and the accumulation of porphobilinogen.
Congenital erythropoietic porphyria • This disorder is due to a defect in the enzyme uroporphyrinogen lll cosynthase. • It is a rare congenital disorder caused by autosomal recessive mode of inheritance, mostly confined to erythropoietic tissues.
Clinical features : • The patients are photosensitive (itching and burning of skin when exposed to visible light) due to the abnormal porphyrins that accumulate. • Increased hemolysis is also observed in the individuals affected by this disorder. • The individuals excrete uroporphyrinogen l and coproporphyrinogen l which oxidize respectively to uroporphyrin l and coproporphyrin l (red pigments).
Porphyria cutanea tarda • This is a chronic disease caused by a deficiency in uroporphyrinogen decarboxylase. • It is the most common porphyria. • It is also known as cutaneous hepatic porphyria. • It is usually associated with liver damage caused by alcohol overconsumption or iron overload.
Uroporphyrin accumulates in the urine. Clinical features: • Cutaneous photosensitivity is the most important clinical manifestation of these patients. • Liver exhibits flourescence due to high concentration of accumulated porphyrins.
Hereditary coproporphyria • This disorder is due to a defect in the enzyme coproporphyrinogen oxidase. • Coproporphyrinogen lll and other intermediates (ALA and PBG) of heme synthesis prior to the blockade are excreted in urine and feces. • Patients are photosensitive. • They exhibit the clinical manifestations observed in the patients of acute intermittent porphyria.
Treatment : • Infusion of hematin is used to control this disorder. • Hematin inhibits ALA synthase and thus reduces the accumulation of various intermediates.
Variegate porphyria • It is an acute disease caused by a deficiency of protoporphyrinogen oxidase. • Protoporphyrinogen IX and other inermediates prior to the block accumulate in the urine. • The urine of these patients is coloured. • Patients are photosensitive.
Protoporphyria • This disorder is also known as erythropoietic protoporphyria. • The disease is due to a deficiency in ferrochelatase. • Protoporphyrin IX accumulates in erythrocytes, bone marrow, and plasma. • Patients are photosensitive. • Reticulocytes and skin biopsy exhibit red flourescence.
Acquired porphyrias • The porphyrias may be acquired due to the toxicity of several compounds. • Exposure of the body to heavy metals (e.g. lead ), toxic compounds (e.g. hexachlorobenzene) and drugs (e.g. griseofulvin) inhibits many enzymes in heme synthesis.
These include ALA dehydratase, uroporphyrin l synthase and ferrochelatase. • Ferrochelatase and ALA dehydratase are particularly sensitive to inhibition by lead. • Protoporphyrin and ALA accumulate in urine.
Diagnosis of porphyrias • To demonstrate porphyrins, UV flourescence is the best technique. • The presence of porphyrin precursor in urine is detected by Ehrlich’s reagent. • When urine is observed under ultraviolet light; porphyrins if present, will emit strong red flourescence.