Download
1 / 41
420 likes | 589 Vues
Forces Involved in Drug-biomolecule Target Interactions: Intermolecular Forces Binding Equilibria. Noncovalent binding equilibria. Binding: noncovalent, reversible association (and dissociation) between molecules

E N D
Forces Involved in Drug-biomolecule Target Interactions: Intermolecular Forces Binding Equilibria
Noncovalent binding equilibria • Binding: noncovalent, reversible association (and dissociation) between molecules • Drug-target complex is more stable (lower in energy) than if the drug is not complexed to the target biomolecule. • Defined rates (kon and koff) and equilibrium constants (Ka and Kd). • Below, AM is the complex; A is the free, unbound small molecule/drug; M is the free, unbound large biomolecule/receptor. • Association equilibrium:
Noncovalent binding equilibria Dissociation equilibrium: Pharmaceutical industry: In general, stronger binding = larger Ka or smaller Kd DG° = -RTlnKeq Useful numbers: 1cal = 4.184J; R = 8.314JK-1mol-1 = 1.9872 calK-1mol-1)
Stabilizing Forces involved in a Drug-Receptor Complex • distance-dependent. • possible when molecular surfaces are complementary. • include (covalent), electrostatic, and hydrophobic interactions. • Covalent bonding • 40-150 kcal/mole. Strongest. • Irreversible: requires a chemical reaction between the receptor and the drug • rare for drug-receptor complexes.
Stabilizing Forces Covalent bonding- example • Example: Anticancer agent 5-fluoro-2'-deoxyuridylate • forms an irreversible complex with thymidilyate synthase • prevents DNA from being biosynthesized • limits the uncontrolled cell division of cancer cells. Ternary Covalent adduct Coenzyme Drug Drug-Target Covalent adduct Target
Stabilizing Forces Electrotstatic interactions (ion-ion; ion-dipole; dipole-dipole including H-bonding; charge transfer; London dispersion forces) Magnitude can be estimated by coulomb’s law (E q1q2/r)) • Ion-ion • 5-10kcal/mole for opposite charges • Ionic compounds have a permanent (full) charge. • Noncovalent (reversible) • Effective over longer distances than other noncovalent interactions.
Stabilizing Forces Electrotstatic interactions, continued • ion-dipole, dipole-dipole • 1-7 kcal/mole • C-Y bonds are polar when Y = an electronegative atom such as O, N, S, halogens • A polar bond leads to partial positive and partial negative charges along the dipole. (Smaller stabilization than full charges) • Relative orientation with respect to the dipole will affect amount of stabilization
Stabilizing Forces Electrotstatic interactions, continued • Hydrogen bond • 3-5 kcal/mole • Special kind of dipole-dipole interaction. • H must be covalently bonded to electronegative atoms N, O, or F • H can interact strongly with lone pairs of heteroatoms. • Optimal geometry - use VSEPR to estimate location of lone pair
Stabilizing Forces Electrotstatic interactions, continued • Cation-pi interactions • 1-3kcal/mole • electron-rich face of aromatic groups plus cationic/electron-poor groups Note: Pi-pi interactions:
Stabilizing Forces Electrotstatic interactions, continued • Induced dipole interactions • Polarization. A charged or polar molecule may induce a dipole in a nonpolar molecule. Very small effects. • Van der Waals or London Dispersion Forces. • ~.5 -1 kcal/mole • Instantaneous dipoles in all molecules stabilize one another. (induced dipole-induced dipole) • Larger complementary surface areas lead to larger London Dispersion Forces.
Stabilizing Forces Hydrophobic interactions Two nonpolar molecules tend to associate in water, due to an increase in the entropy of water molecules
Additional Structural Considerations 1. pH/pKa and drug-target interactions. The protonation state of a particular functional group will determine its charge, and therefore the nature of intermolecular forces 2. Stereochemistry and drug-target interactions. Different stereoisomers can have different activities. (Not equally complementary to the 3D structure of the target). Ex 1. R and S isomers of the antimalarial chloroquine have equal potencies:
Additional Structural Considerations Ex. 2. the 1R, 2S enantiomer of norephedrine (2-amino-3-phenyl-1-propanol) is 100 times more potent than the 1S,2R enantiomer on the alpha adrenoreceptor in vivo and in vitro. Ex. 3 S-Ketamine is an anaesthetic; R-ketamine has little anaesthetic action but is a psychotic.
Additional Structural Considerations • 3. Conformation and drug-target interactions. • Both drug and target molecules may have multiple conformations. • Recall Morphinan from Molecular Conceptor in lecture 1 Drugs can have higher potency if they are "conformationally restricted" to a bioactive conformation. Bulky substituents or rings are often used for this purpose:
Stabilizing Forces - Summary Keq = Ka A + M AM DG° = -RTlnKeq DG° = DH° - TDS° Electrostatic interactions Enthalpic (DH) effects. Placement of complementary groups on drug and target. Size of charges, distance between interacting groups, orientation. Multiple small effects add up! Hydrophobic interactions, conformational restriction Entropic (DS) effects. A + M AM DG° Free energy of Binding of drug to target A + M AM Reaction coordinate
Hydrophobicity (lipophilicity) and drug action. • Hansch and coworkers hypothesized two steps for a drug to work: • Pharmacokinetics (drug getting to the site of action) • Pharmacodynamics (interaction of drug with the site). • Pharmacokinetics phase depends on interaction with aqueous AND membrane environments. • The ability to interact with nopolar membrane environments can be correlated with a water-octanol partition coefficient P.
Hydrophobicity (lipophilicity) and drug action. • Note optimum partition coefficient: • if a compound is too hydrophobic, it will remain in the first membrane it contacts; • if it is too hydrophilic, it will never cross cell membranes to get to its site of action.
Predict Possible binding interactions with targets Protein kinase inhibitors: bind in pocket where ATP binds
Predict Possible binding interactions with targets • Authors note: • N1 H-bond with leu 83 amide NH • ArOH H-bond with Asp 145+Lys 33** • ArOH edge to face with Phe80 • ArOH hydrophobic pocket Bound to CDK2
Predict Possible binding interactions with targets • Authors note: • N1 H-bond with met109 amide NH • N3 H-bond with water that H-bonds with Thr106 (no room in CDK2with Phe80 • ArSCH3 in pocket • Quinazoline in hydrophobic pocket Bound to p38
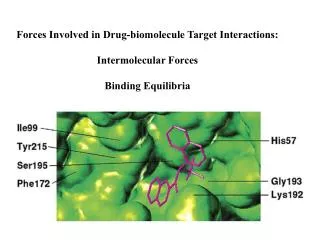
Predict Possible binding interactions with targets Nonpeptide inhibitors of serine protease cathespin G (associated with inflammation) identified by high-throughput screening of a diverse library of compounds.
Predict Possible binding interactions with targets • Authors note: • Pi stacking of 2-naphthyl with his 57 • P-OH H-bonded to His 57 • P-OH H bonded to amide NH of gly 193 = • “oxyanion hole” of serine proteases • P-OH H bonded to NH3 of lys 192 • Ketone H-bonded to lys 192 IC50 = 4 mM
Crystal structure showed hydrophobic residues: phe 172, tyr 215, Ile 99. Can “fill” this hydrophobic pocket: New inhibitor designed… IC50 = .053 mM for R =
Drug Targets - an Overview • Lipids • Carbohydrates • Proteins • Carrier proteins • Enzymes • Receptors • Nucleic Acids
Drug Targets: Lipids • Few drugs interact with lipids • They often act by disrupting lipid structure of cell membranes. • Ex 1. General anaesthetics. • Ex 2. Amphotericin B (used to treat athletes foot) binds to fungal cell membranes, creating channels and killing fungus. Preferentially binds to ergosterol (in fungal membranes) over cholesterol (in mammalian membranes).
Drug Targets: Carbohydrates • energy sources • structural elements in the cell • involved in specific binding interactions between receptors and ligands. Ex 1. Influenza virus binds to its host by a cell surface sugar and sialic acid residue - a drug that binds more strongly than the natural binding site will block the viral attachment. Ex 2. Doxorubicin (anticancer agent) linked to a carrier with a specific carbohydrate is more efficient at killing colon cancer cells than doxorubicin administered alone.
Drug Targets: Proteins - Carrier proteins/transporters Ex. Fluoxetine (prozac) works by binding to the transporter for the neurotransmitter serotonin, preventing its uptake into the cell.
Drug Targets: Proteins - Enzymes • Enzymes are a major target for drugs. • Enzyme targets of microorganisms, viruses used to fight infection • The body's own enzymes can be targets (if there is an excess or deficiency of a metabolite). • A drug may act by binding • strongly but reversibly to the active site (competetive inhibitor), • reversibly to a different site (allosteric inhibitor), • irreversibly to the active site. • The affinity of inhibitors is determined by enzyme kinetics. Review any biochemistry text for details of this analysis.
Drug Targets: Proteins - Enzymes Ex. 1 Adenosine deaminase metabolizes adenosine and degrades many antiviral and cancer therapy treatments. Inhibitors will help make those drugs more effective. A drug that resembles the transition state of the catalyzed reaction should bind very strongly to the enzyme active site, improving the effectiveness of other therapies. Reaction: Drugs:
Drug Targets: Proteins - Enzymes Ex 2. Tetrahydrofolic acid is necessary for the synthesis of nucleic acids. Bacteria must synthesize it to survive (humans ingest it). Tetrahydrofolic acid The drug Prontosil was found to be bacteriostatic. Prontosil is a prodrug, because it is metabolized to form the actual active agent p-aminobenzene sulphonamide (sulfanilamide). It resembles the structure of the substrate p-aminobenzoic acid (above), so it will bind to the active site of the enzyme dihydropteroate synthase.
Drug Targets: Proteins - Enzymes Ex. 3: antihypercholesterolemic drugs. Cholesterol: major component of fatty plaque deposits on inner wall of arteries, and ~50% is synthesized in the body. Hypercholesterolemia is a primary risk factor for coronary heart disease. Therapeutic goal: decrease the amount of cholesterol synthesized in the body. The rate-determining step is the following, catalyzed by HMG-CoA reductase: HMG-CoA
Drug Targets - Proteins - Enzymes HMG-CoA: Km = 10-5M Hydrolysis product Mimics HMG-CoA R=CH3: mevinolin lovastatin KI = 6.4x10-10M R=H: compactin; KI = 1.4x10-9M
Drug Targets: Proteins - Receptors • Major target for drugs • Receptors are used by cells for communication • In nerve cells, electric impulses are "communicated" to cells through a chemical message (neurotransmitter) that is received by a protein receptor embedded in the cell membrane. Binding of this neurotransmitter results in a biological response • Other chemical messages are hormones that are circulated through the body. They also bind to specific receptors, triggering a biological response.
Drug Targets: Proteins - Receptors • Two main mechanisms to transmit the message from the outside of the cell (hormone or neurotransmitter messenger) to the inside of the cell (second messenger): • ion channels • membrane-bound enzymes. • Drugs may be agonists or antagonists that control the activity of receptors • Agonists act like natural messengers. To design a drug agonist, the starting point is the natural ligand. • Antagonists block the receptors from the natural messenger. To design a drug antagonist, the structure is not generally similar to the natural ligand. Ideally, the structure of the receptor would be a good starting point. If unknown, use information about any antagonists or even agonists as a starting point. • Ex.: cimetidine (Molecular conceptor , lecture 1) is a histamine receptor antagonist
Drug Targets: Nucleic Acids Drugs that interact with DNA are usually very toxic because human DNA and pathogen DNA are very similar. For cancer treatment, the only difference between cancer cells and normal cells is the rapid cell division. Therefore, drugs that halt mitosis (DNA synthesis) should preferentially halt the mitosis of cancer cells. Ideally, a drug would be able to bind to specific sequences.
Drug Targets: Nucleic Acids Three main classes of drugs that interact with DNA: 1. DNA intercalators. Bind reversibly between the base pairs. Disrupt DNA structure and prevent normal functions of DNA.
Drug Targets: Nucleic Acids 2. DNA alkylators. Form covalent bonds (irreversible) with DNA. Nucleophile = N, O, S atoms in DNA that aren't sterically hindered or involved in H-bonding. Electrophile = alkylating agent. Anchimeric assistance DNA Interstrand crosslink. 3. DNA strand breakers. Many complex reaction mechanisms are being uncovered, but when these drugs bind to DNA, the result is strand breakage.
Diversity of Targets; Diversity of Rationales This was an overview of main targets of drug action. Each general target could take weeks of classtime to discuss! For your presentations/papers, you may have to research pertinent details of the target or drug action… References: Anslyn, E. V.; Dougherty, D. A. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2004. Thomas, G. Medicinal Chemistry An Introduction; John Wiley & Sons: New York, NY, 2000. Patrick, G. L. An Introduction to Medicinal Chemistry; Oxford University Press: New York, 2001. Silverman, R. B. The Organic Chemistry of Durg Design and Drug Action; Academic Press: New York, 1992. Shewchuk, L; Hasssell, A.; Wisely, B.; Rocque, W.; Holmes, W.; Veal, J; Kuyper; L. F. J. Med. Chem.2000, 43, 133-138.