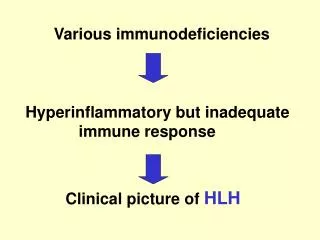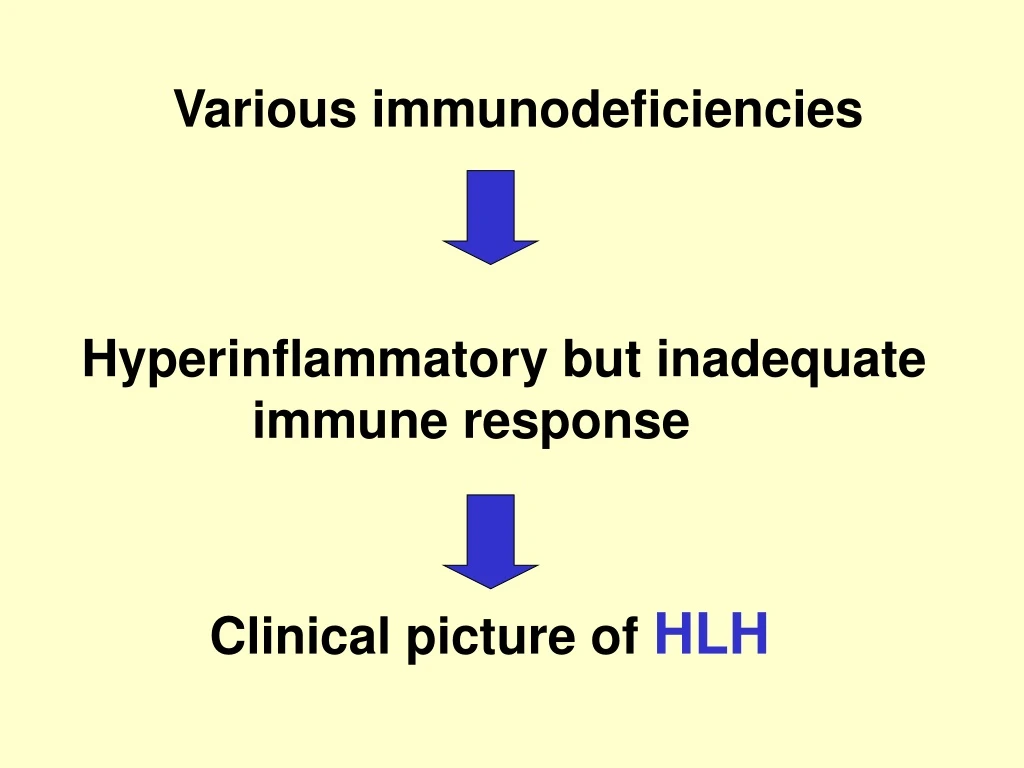
Various immunodeficiencies Hyperinflammatory but inadequate
120 likes | 132 Vues
HLH is a hyperinflammatory syndrome characterized by inadequate immune response. Clinical features include high fever, hepatosplenomegaly, pancytopenia, and neurological symptoms. Diagnosis is based on criteria such as hemophagocytosis and hypercytokinemia. Treatment options include cytostatic drugs and bone marrow transplantation. Prognosis varies, with relapse-free survival reported in 60-70% after BMT.

Various immunodeficiencies Hyperinflammatory but inadequate
E N D
Presentation Transcript
Various immunodeficiencies Hyperinflammatory but inadequate immune response Clinical picture of HLH
Hemophagocytic lymphohistiocytosis (HLH) Clinical symptoms and findings High fever, hepatosplenomegaly, pancytopenia Neurological symptoms, jaundice, edema, lymphadenopathy, rash High triglycerides, low fibrinogen, coagulopathy high ferritin, transaminases, bilirubin, LDH CSF pleocytosis and/or elevated protein Hemophagocytosis in BM or other organs
HLH Variable course of disease Rapidly progressive leading to death within weeks Transient improvements with unspecific therapies Disappearance of symptoms without therapy Disappearance of symptoms with immuno- suppressive/immunomodulatory drugs
HLH Classification Genetic, primary HLH Acquired, secondary HLH FHLH - Perforin mutations (chr.10) Exogenous agents - infectious organisms, toxins - Chromosom 9 linkage (VAHS, IAHS) - Unknown mutations HLHEndogenous products - tissue damage - radical stress - Immune deficiencies - metabolic products CHS Griscelli syndrome Rheumatic disorders XLP Malignancies SCID GJ 2002
„IAHS“ in Childhood (219 cases from the literature) 1979-1995 Organism Clinical outcome Dead Alive No data EB Virus 121 72 27 22 Other viruses 28 11 13 4 Bacteria 11 2 9 0 Fungi 2 1 1 0 Protozoae 1 0 1 0 No organism 57 13 33 11
„IAHS“ in Childhood (219 cases from the literature) (1979-1995) Age Clinical outcome Dead Alive No data < 3 years: 77 40 26 11 > 3 years: 82 29 47 6 „Children“: 60 60 22 4 Total 219 103/198 95/198 (52%) (48%)
HLH Diagnostic criteria Histiocyte Society 1991 Clinical Fever > 38.5 Splenomegaly Laboratory Cytopenia of => 2/3 cell lines Hypertriglyceridemia and/or hypofibrinogenemia Histopathology Hemophagocytosis in bone marrow or spleen or liver or lymphnode Strong supportive evidence are spinal fluid pleocytosis, liver histology resembling chronic persistent hepatitis, low natural killer cell activity
HLH Immunological parameters Hypercytokinemia (TNF, INF, IL 6, IL 8, IL 10), Increased soluble CD 25 (interleukin 2 receptor -chain) Increased soluble CD95-ligand NK-cell activity below 5% lysis CD2/CD86 positive cells in lymphocyte gate Phagocytosing dendritic cells in culture
HLH Therapy Cytostatic and immunsuppressive/ immunomodulatory drugs: Corticosteroids, Cyclosporin A, Etoposide Immunoglobulins, Antithymocyte globulin Bone marrow transplantation Prognosis In 20% no response to therapy After BMT 60-70% relapse-free survival