Drug Development in Latin America Recent Trends and Competitive Advantage
Drug Development in Latin America Recent Trends and Competitive Advantage. Key Objectives. Overview of Clinical Drug Development in the region General demographic factors in the region Shift from traditional regions to emerging markets and clinical trial saturation


Drug Development in Latin America Recent Trends and Competitive Advantage
E N D
Presentation Transcript
Drug Development in Latin America Recent Trends and Competitive Advantage
Key Objectives • Overview of Clinical Drug Development in the region • General demographic factors in the region • Shift from traditional regions to emerging markets and clinical trial saturation • Potential pros and cons from the pharmaceutical industry perspective • Generic regulatory aspects of EC and agency communication: • Generic regulatory flow • Communication with EC and agencies • Understand country specifics: • Focus on Mexico, Brazil, Colombia, Peru, Chile and Argentina • Overview of regulatory steps, timelines and comments • Focus on recent updates and challenges
Drug Development in Latin AmericaPharmaceutical Industry Perspective Carlos Caparros, MD Sr.Director Global Clinical Operations Latin America-Schering Plough
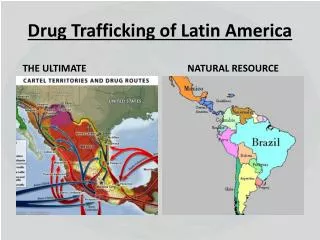
Latin America-Characteristics • Population 500 + million • 300 + m Spanish speaking • 190 m (Brazil) Portuguese speaking • Mainly located in large cities • Large Metropolitan areas • Large Paediatric population • +130 m aged 0-14 years (30%) • All key countries with local regulations aligned with Declaration of Helsinki, CIOMS and ICH-GCP guidelines • All key countries with a regulatory agency that regulates drugs, devices and clinical research (usually a branch of the Countries Ministry of Health-MOH of each country). • All key countries function with a mix of local and central IRBs.
Latin America-Characteristics • Fast enrollment of patients • Competitive costs • Convenient time zone to interact with the US and Europe • Investigators and patients eager to be part of clinical trials • 50% of the pharmaceutical growth by 2020 is coming from emerging regions (15-18% from LA) • Quality standards comparable to other “traditional research markets”
Latin America-Demography Ref: *Spanish speaking, no difference for research purposes (one protocol, one label for all); #Portuguese speaking.
Clinical Trials in Latin America (n: +4100 trials) Source: Clinicaltrials.gov April 2009
Saturation of Clinical Trial Sites Source: Raps Focus, 2009, Latin American CTAs
FDA Regulated Investigators Source: Raps Focus, 2009, Latin American CTAs. FDA regulated investigators 1996 vs 2006
Key Regulatory Players SITES LATAM Representative (Affiliate or CRO) SPONSOR (ex-LATAM) ECs & MOH
Clinical Trial Application Process CTA Preparation CTA Submission EC & MOH Follow Up CTA EC & MOH Approval Queries
Latin America Generic Regulatory Flow Protocol, ICF, IB, POA, Insurance Drug at Depot Time to Submission 2-4 weeks Import process 1-8 week Ethics Committee (EC): -IEC + Site: ARG -IEC: PER -Regional: CHI -Site: MEX, COL, BRA Import License Time to IL 1-8 week MoH Submission MoH Approval Time to 1st EC Approval 4-10 weeks Time to MOH Approval 4-30 weeks
Clinical Trial Application Regulatory Queries Ref: 1-COL & MEX. 2-CHI & PER. 3-BRA & ARG
Pitfalls in Clinical Trial Application Process • Clear up front understanding of steps and requirements with involvement of regulatory and start up groups from the start (RFP>Kick off>Start Up phase) • Country specific regulatory plan and strategy • Best & Worse scenario timelines, taking into account not only Final Protocol to FSI, but Final Protocol to Enrollment Commitment Achieved. • Communication and Follow up plan in the CTA process (Who, When, How) • Definition of query resolution process for each of the different type of queries • Strict timelines in all the non rate limiting steps • Strategic follow up and fast query turn around time in the rate limiting steps
Drug Development in MexicoRecent Updates and Challenges Mirtha Lipezker, MD Sr.Director Global Clinical Development & Country Manager-Kendle Mexico
Mexico Flow Chart Translation of docs. 2-3 weeks Commercial Invoice content is reviewed with sponsor Regulatory docs. from sites E.C., Hospital, study staff 4-6 weeks Protocol Submission to MOH MOH (COFEPRIS) Approval 6-8 weeks Total Approval & Set Up Process 13-17 Weeks Import license (not required for study drug or lab kits) 2-3 weeks Customs release 1-2 days DEPOT
Mexico Comments • The COFEPRIS* regulates clinical trials in Mexico. • Key Initial documents: Protocol, ICF, IB • Study documents (except investigator brochure) in Spanish • Local regulation requests Ethics Committees (EC) to approve the protocol per site. With the first site’s EC approval and the sites director authorization letter the protocol can be submitted to the MoH for clinical trial authorization. Some sites might have a scientific committee and an ethics committee, approval by both is required. • The process is sequential, additional sites must be approved by the MoH (4 weeks). • Insurance certificate endorsed by local insurance company needed before site activation * COFEPRIS – Comisión Federal para la Protección Contra Riesgos Sanitarios
Mexico - Updates and Challenges • Updates to Local Regulations: • Custom authorities have removed the requirement for import license for drugs or for export license on blood serum samples. This has reduced the start up timeline from 3.5-4.5 months to 3-4 months, being Mexico figures in line with some EU countries • Challenges: • Social Security hospitals (IMSS) require a national committee approval (Comisión Nacional de Investigación Cientifica) • Negotiation of contracts in this sites is more complex than in other private institutions
Drug Development in BrazilRecent Updates and Challenges Alcione Braga, RPh, MBA Director Global Clinical Development & Country Manager-Kendle Brazil
Brazil Timelines * It is necessary 1 week to prepare the ANVISA dossier after receiving the local EC approval.
Brazil Flow Chart Time to documents translation into Portuguese after all required documents arrival from sponsor and dossier preparation before submission: 3 -5 weeks Kendle Total Set Up & Approval Process: 30-41 weeks Investigator MoH (ANVISA) Local Ethics Committee Time to IRB/EC approval: 5 -8 weeks MoH submission after getting the Local IRB/EC approval: 1 week Time to MoH approval: 18 - 23 weeks National Ethics Committee (CONEP) Import Product Time to CONEP approval: 18 - 23 weeks Time to import: 3 - 4 weeks Study Start
Brazil Comments • The ANVISA (Agência Nacional de Vigilância Sanitária) regulates clinical trials in Brazil. • Key Initial Docs: Protocol, ICF, IB, POA, Protocol approval letter from an IRB of the origin country (e.g. where the sponsor company is located), Insurance applicable to Brazilian sites. • Other Study docs: Sponsor statements: Treatment & Assistance; Local Regulations; Biological samples; Study results publication; Study termination; Access to medicine). • Study documents (including investigator brochure) in Portuguese • Three steps for clinical trials: CEP (local EC), CONEP (National EC) and ANVISA (MoH). CONEP and ANVISA are parallel process for part of the process. • Insurance certificate needs to cover specifically Brazil and preferably its sites • A list of the study drug and material to be imported during the whole study period needs to be submitted to the MoH.
Brazil – Updates and Challenges • Updates: • Resolution 39 went into effect on 5 July 2008. • Made the national EC (CONEP) and CA (ANVISA) reviews for approval a truly parallel process as in EU: • Potential savings of 6-8 weeks for first site (Coordinator Site) to reach SIV. • Granted competent authority (ANVISA) ability to approve all study sites in one submission and review cycle. • Saving 4-6 week for subsequent sites to reach SIV. • Importance: If law is effective, it will reduce approval time in BR from 10 to approximately 8.5 months (Assuming no questions from CONEP or ANVISA). • Challenges • Protocols with placebo arm are difficult to obtain CONEP approval, if no strong medical and ethical rationale to justify it. • Nowadays, patients access to medication after the trial is one of important CONEP’s concerns.
Drug Development in PeruRecent Updates and Challenges Jacqueline Zeuner, MS Director Global Clinical Development & Country Manager-Kendle Peru
Peru Timelines *Add 4 wks for Biological
Peru Flow Chart Translations: 2 weeks • MoH submission and evaluation of: • Clinical trial (INS) • Investigational product (DIGEMID) • 40 working days for non biological products • 60 working days for biological products Time to MOH Approval 10-14 weeks Regulatory documents Protocol IB, ICF, POA, Insurance 6 weeks for Central E.C. approvals and study site approvals MoH Approval (INS/DIGEMID) valid for 1 year Total Approval & Set Up Process 20-27 Weeks Import Licenseby DIGEMID: 3 weeks Customs release: 4 days DEPOT
Peru Comments • The INS (Instituto Nacional de Salud) regulates clinical trials in Peru. As of July 29, 2006, trials are also subject to state law. • Key Initial Docs: Protocol, ICF, IB, POA, Insurance policy • Other docs: Hospital Directors letter, drug label, list of materials to be imported • Study documents (including investigator brochure) in Spanish, • In order to perform clinical trials in Peru, Sites, Ethics Committees (ECs), and CROs, must be registered with the INS. The INS keeps a register of approved sites, CROs and ECs, which is available on their website or by request. • Central EC approvals for all participating sites have to be obtained prior to MoH submission (by one of the EC’s approved by the MoH) and also by Local site EC (if applicable) • A list of the study drug and material to be imported during the whole study period to be submitted to the MoH. • Process is sequential, but additional sites or amendments can be added once the original MoH approval has been obtained. • Import license is granted by DIGEMID (Dirección General de Medicamentos, Insumos y Drogas) with strict review of supply request, COA and labels. • Free contraceptives need to be provided throughout the study. Local logistic preferred versus importation.
Peru - Updates and Challenges • Updates to Local regulations: INS will become more strict regarding: - Number of clinical studies per Principal Investigator - Therapeutic experience of PI and staff - Local endorsement of clinical trial insurance • Challenges:INS is conducting more frequent inspections
Drug Development in ColombiaRecent Updates and Challenges Mariagabriela Alterio, MS Director Global Clinical Development & Country Manager-Kendle Colombia
Colombia Flow Chart Total Approval & Set Up Process 17-21 Weeks Protocol, ICF, IB, Insurance, POA Drug at Depot Time to Submission first site 2-3 weeks Import process 1 week Site IRB’s Import License Time to IRB Approval 4-5 weeks Time to IL 2 week MoH (INVIMA) Submission MoH Approval Time to MOH Approval 8-10 weeks
Colombia Comments • The INVIMA (Instituto Nacional de Vigilancia de Medicamentos y Alimentos) regulates clinical trials in Colombia. • Key Initial Docs: Protocol, ICF, IB, POA, Insurance policy for Colombian sites • Study documents (except investigator brochure) in Spanish • Local regulation requests Institutional review boards to approve the protocol per site. With the first site’s IRB approval the protocol can be submitted to the MoH for approval. • Additional sites must be notified to the MoH. • Insurance policy needed to cover all Colombian sites • With MoH approval the import license for study drug and other supplies (kits) may be requested. • No special issue with placebo controlled studies if approved by ECs.
Colombia - Updates and Challenges • Updates to Local regulations: • Resolution 2378 / 2008: GCP and certification of institution by MOH to be able to perform clinical research. MOH will certificate sites • Certification needs to be completed in 2 years and will be valid for 5 years. • Challenges: • New resolution requested all investigational sites to present certification plan and timelines to the MOH by 26 Dec 2008 • MOH will do an inspection to the sites as needed • All protocol submission to the MOH for approval will need to include the copy of the submission of a certification plan.
Drug Development in Argentina/ChileRecent Updates and Challenges Raul Bozzo, MD Director Global Clinical Development Kendle Argentina-Chile
Argentina Timelines *Add 2-4 wks in phase II, vulnerable populations, complex designs
Argentina Chart Total Approval & Set Up Process 25-29 Weeks Protocol, ICF, IB, Power of attorney, Agreement (POA) Drug at Depot Time to Submission 2-3 weeks Import process 1-2 week Independent Ethics Committee (IEC) Import License Time to IEC Approval 2 weeks Time to IL 1-2 week Site IRB’s or Equivalents MoH (ANMAT) Submission MoH Approval Time to 1st IRB Approval 2 weeks Time to MOH Approval 17-18 weeks
Argentina Comments • The ANMAT (Administracion Nacional de Medicamentos y Alimentos) regulates clinical trials in Argentina. • Key Initial Docs: Protocol, ICF, IB, POA, Contract template • Other docs: Ministry of Justice approval of ICF; Sites director approval letter and insurance • Study documents in Spanish • A central IEC approval plus site IRB approval have to be obtained prior to MoH submission for a lead site. MoH approval for other sites can be obtained by fast track (4weeks). • Insurance certificate endorsed by local insurance company needed before site activation • A list of the study drug and material to be imported during the whole study period needs to be submitted to the MoH.
Argentina - Updates and Challenges • Updates to Local regulations: • 2008 Updates in ANMAT 5330/97 regulation: Expands requirements on documents for Clinical Trial Application´s (Institution Authorization, Locasl Health Notification and ICF) • ANMAT Approval (1310/09) of new GCP guidelines (1490/07) for Argentina following the spirit of ICH and The Document of Americas PAHO 2005 • Challenges: • MOH Approval for protocols with placebo arm when standard treatment is available: • Add on design (example: placebo controlled add on to metformin in t2d) • Approved non pharmacological initial therapy according to clinical guidelines (example: dyslipidemia or hypertension) • Medical and ethical rational (according to clinical guidelines) • Clearly defined bail out or rescue criteria or possibility to roll over to active or open label treatment
Chile Timelines * Delays can be possible depending on the volume of work of each CEC
Chile Flow Chart Total Approval & Set Up Process 14-21 Weeks Protocol, ICF, IB, Insurance, POA Drug at Site Time to Submission 2-3 weeks Time to drug at site 1 week Regional EC / IRB’s Custom Destination Time to Regional EC/IRB Approval 6-10 weeks Time to CD 1 week (or less) MoH (ISP) Submission MoH Approval Time to MOH Approval 4-6 weeks
Chile Comments • The ISP (Instituto de Salud Pública) regulates clinical trials in Chile (MoH). • Key Initial Docs: Protocol, ICF, IB, POA, Insurance • Other docs: Sites or Hospital director approval letter • Study documents (including investigator brochure and contract) in Spanish. • Only one regional EC (CEC: Comite de Etica Cientifico) approval has to be obtained prior to MoH submission in order to obtain drug import authorization. The rest of EC approvals have to be submitted to the MoH before receive study drug. • Some sites have also the institutional IRB that also needs to approve the protocol. • For vaccine studies approval of the Extended Program on Immunization (EPI) is needed. Submission can be in parallel to the regional ethics submission. • Local Insurance Certificate or local representative company endorsement will be needed. • A list of the study drug to be imported during the whole study period is need to be submitted to the MoH. • In placebo controlled studies a placebo justification letter will be needed.
Chile - Updates and Challenges • Updates to Local regulations: According to a new MoH Resolution (following spirit of Document of Americas-PAHO) to be released soon. • Insurance Policy with Local Representation starting to be required for MoH submission. • Authorization Letter from Director of the Institution dated after Ethic Committee approval date, is starting to be required for MoH submission. • Minimum requirements for label format. • Local SAEs starting to be submitted in 5 working days. • Monitor(s) CVs starting to be required for MoH submission. • Challenges: • MoH has started to perform Site Audits from 2008. • Trained and available staff need to be demonstrated prior to MoH submission. • Several new MoH requirements are not coincident with Ecs requirements. MoH - ECs (Health Services) meetings are planned to improve points of action and processes.
Summary • Six Latin American countries account for more than 2/3 of the regions population and approx. 70% of the trials. • Shift in worldwide clinical research to emerging markets like Latin America • Proactive up front involvement, communication and planning of customized regulatory strategy follow up and query reply are key for CTA success • Latin American country regulations in all key countries are in a phase of growth and development: • Investigator, Sites and IEC/IRB training and requirements: better protection of subjects and more quality data • Facilitate certain steps in the regulatory flow (Mexico, Brazil) that will shorten regulatory timelines • Increase in number of MOH inspections: more local control and improvement in quality and assurance • Common regional characteristics, high enrollment rates, lack of saturation, competitive costs and growing regulations predict continuous growth of clinical research in the region