Stratton Nature 45: 719 , 2009
210 likes | 438 Vues
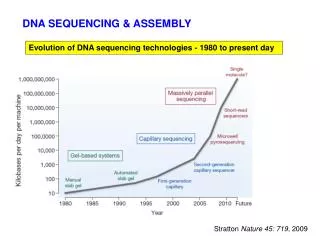
DNA SEQUENCING & ASSEMBLY. Evolution of DNA sequencing technologies - 1980 to present day . Stratton Nature 45: 719 , 2009. $$$ Motivation to “spur DNA sequencing technologies, boost accuracy and drive down costs” .

Stratton Nature 45: 719 , 2009
E N D
Presentation Transcript
DNA SEQUENCING & ASSEMBLY Evolution of DNA sequencing technologies - 1980 to present day Stratton Nature 45: 719, 2009
$$$ Motivation to “spur DNA sequencing technologies, boost accuracy and drive down costs” “The X Prize Foundation of Playa Vista, California, is offering a $10-million prize to the first team to accurately sequence the genomes of 100 people aged 100 or older, for $1,000 or less apiece and within 30 days [beginning September 5, 2013].” …with an accuracy of <1 error per 1 million bases see Nature 487:417, July 26, 2012
Sanger chain termination method (Fred Sanger, 1977) - enzymatic synthesis of DNA strand complementary to “template” of interest Nobel Prizes: Sanger 1958 (protein structure) 1980 (DNA sequencing) … but it stops when dideoxynucleotide is incorporated 4 parallel sets of reactions: ddATP + 4 dNTPs ddCTP + 4 dNTPs etc. Fig. 4.2
Ratio of ddATP:dATP important to get appropriate size range of products - set of products each terminating with ddA - their sizes reflect positions of T in template DNA Fig. 4.2
Products (each differing in length by 1 nt) resolved on denaturing polyacrylamide gels... or by capillary electrophoresis … Fig. 4.1 Autoradiograph Automated sequencing profile Fig. 4.3
PRIMERS FOR SEQUENCING 1. “Universal” - forward & reverse 2. Custom-designed “internal” Fig. 4.5 • use new sequence info to design primer to sequence next stretch If insert is too long to completely sequence using “universal primers”, can use this strategy to close a “sequencing gap”
… or can find another clone in library that has overlap, and sequence it using “universal” primers Fig. 3.35
What if there is a “physical gap”? if particular region of genome is not represented in clone library Fig. 4.17 • can use a different vector to prepare a second clone library (maybe region was unstable in first vector) • then use probes (eg. oligomers) mapping to ends of contigs from • first library to screen second library
Example of closing a “physical gap” 8 7 1 2 Fig. 4.11 You have 9 contigs & design oligomers mapping close to their ends (#1-18) screening by hybridization … or byPCR Which contigs are adjacent?
What if “physical gap” is very short? < 10 kb or so - could use oligomers mapping to ends of contigs in PCR reactions with uncloned DNA template … 3’ 5’… 3’… … 5’ - then sequence the PCR product directly - this slide also illustrates a method for finding overlapping clones
ASSEMBLING INFO FROM CLONES INTO CONTIGS 1. CHROMOSOME WALKING by hybridization - sequence from one clone is used as probe to screen library of clones to find overlapping one - repeat to “walk along” genome Fig. 4.12
But what if probe contains repeated sequences? - so hybridizes to multiple clones Problem avoided if use short unique-sequence probe (eg oligomer) mapping close to end of clone … or if pre-hybridize with repeat sequence Fig. 3.34
2. CHROMOSOME WALKING by PCR Fig. 4.13 - design primer pairs based on sequence at end of clone - use other clones in library for template DNA - will get PCR amplicon for any new clones with that sequence - reactions can be carried out as pools for more rapid screening (combinatorial screening)Fig. 4.14
3. CLONE FINGERPRINTING To identify overlapping clones: by finding features that they share Restriction profile fingerprint or clones having STS in common (Fig.4.15D) Fig. 4.15A
Haemophilus genome project 1995 (1.8 Mbp) 1. DNA sonicated, fragments (1.6 – 2 kb) cloned in plasmid vectors 2. Shotgun sequencing of insert ends ~ 20,000 clones analyzed, 11 Mbp of sequence, scaffolds with sequencing gaps & physical gaps 3. Assembled into 140 contigs 4. Screened for overlapping clones – reduced to 42 contigs 5. Assumed gaps represented genome regions unstable in plasmid vector - switched to lambda vector 6. Probed l library with oligomers from contig ends or used PCR with primer pairs from contig ends Fig. 4.10
“Next generation” sequencing technologies “Cost per Megabase of DNA Sequence (or Why biologists panic about computing)” - major challenge to correctly assemble the massive amount of sequence data generated… and to interpret it ! National Human Genome Research Institute
1. Pyrosequencing C Genome Res 11:3, 2001 Fig. 4.9 - one dNTP is added at a time + enzyme (apyrase) that degrades dNTP if not incorporated into new strand, then next dNTP added - incorporation detected by chemiluminescence of pyrophosphate (PPi) www.youtube.com/watch?v=kYAGFrbGl6E&feature=related
“Massively-parallel”pyrosequencing (on beads or chips) Polymerase Enzymes on beads and primer 454 technology PPi Genomic DNA PCR Light Sample preparation Pyrosequencing - DNA sheared, adaptors ligated, attached to bead & PCR amplified - beads captured in wells & pyrosequencing carried out in parallel on each DNA fragment - average read of ~ 700 (?) bp ... but “up to 1.6 million reactions can be carried out in parallel on a 6.4 cm2 slide” “expect ~ 500 million nucleotides of sequence data per 10 hour run” (July 2010) Medini Nat Rev Microbiol. 6:419, 2008
Sequencing by synthesis Sample preparation 2.Illumina sequencing (parallel microchip) SOLEXA technology - add adaptors to sheared DNA, attach to chip, then PCR “bridge amplification” - denature clusters of ~ 1000 copies of DNA molecules & sequential sequencing using four fluorophore-labelled nts HiSeq 2000 HiSeq 1000 - average read of ~ 40-100 bp (short-read) (Illumina website Sept. 2012) www.youtube.com/watch?v=HtuUFUnYB9Y&feature=related Medini Nat Rev Microbiol. 6:419, 2008
3.Single molecule real-time sequencing (Helicos, Pacific Biosciences) - nanoscale wells on chip so ~ one DNA polymerase molecule per well - continuous monitoring of nt incorporation (rather than termination as in Sanger method…) and no amplification - formation of phosphodiester bond releases fluorophore - read length 25 to 55 bases, 21-35 Gigabases per run Metzker Nature Reviews Genetics 11:31, 2010 (Helicos website Sept. 2012)
Press release, Dec 9,2010: “PacBio & Harvard Use Fast Gene Sequencer to Crack DNA Code of Haitian Cholera Strain” Chin et al. New Eng J Med 364:33, 2011 H1 and H2 strains were sequenced in < 24 hr with enough “reads” to cover the genomes 60 and 32 times, respectively.