Download

1 / 50
530 likes | 946 Vues
Sequencing the Maize (B73) Genome. Maize Genome Sequencing Consortium. G enome S equencing C enter. The Plan. Progress as of 9/30/06. BAC-by-BAC Strategy to Sequence the Maize Genome. Maize B73 Genome (2300 Mb). BAC library construction

E N D
Sequencing the Maize (B73) Genome Maize Genome Sequencing Consortium Genome Sequencing Center
BAC-by-BAC Strategy to Sequence the Maize Genome Maize B73 Genome (2300 Mb) BAC library construction (Hind III, EcoR I/MboI ; 27X deep ; 150kb avg. insert) Genetic Anchoring in silico, overgo hybridization Fingerprinting ~460,000 BACs BAC End Sequencing ~800,000 BAC physical maps (HICF & Agarose) FPC databases (Agarose and HICF) STC database Choose a seed BAC Shotgun sequencing and finishing STC database search, FP comparison Determine minimum overlap BACs Complete maize genome sequence
Map Summary • Total Assembled Contigs: 721 • Equal to 2,150 Mb, 93.5% coverage of 2300 Mb genome • Anchored: 421 ctgs, 86.1% the genome • average anchored contig size: 4.7 Mb • Unanchored: 300 ctgs, 7.4% coverage average unanchored contig size: 0.56 Mb • 189 of the 300 unanchored contigs are less than 10 clones • Largest anchored contig 22.9Mb in Chr9 • Largest unanchored contig 6.7 Mb • Total FPC Markers: 25,924 • STS markers: 9,129 • Overgo Markers: 14,877 • Anchored markers: 1918
MTP Selection • Seed BACs: 4000, done • Mega Contig: 197, done • Clone Walking from Seed BACs: 2,800 done; in progress • Total clones picked = 6,997 • On track to deliver 1000 clones/month until maze MTP is complete
Flowchart for MTP picking and Library Construction Clone selection (combine seed BAC and BAC end sequences with fingerprinting and trace files) Clone picking (Resource Center) GenBank BAC end sequence database MTP sequencing Seed BAC database Library DNA production Library DNA production DNA shearing Hfq sequencing MTP BAC end database Clone verification Clone shipping Continue shotgun library construction at WashU
Seed BAC Walking In Agarose and HICF map, selecting large clones next to seed BAC Blastn search of BAC end sequences against seed BAC sequences Check blastn alignment for candidate clones Check trace file for Dye blob Check the Sulston score in HICF map for overlap Check Agarose fingerprints to avoid overlap with large bands Choose walking clone
Minimum Tile Path Pipeline • BAC End Sequence of potential BACs are BLASTed against the Seed BACs • Results are classified based on location on the FPC • A table for each BAC is created of filtered BLAST results with links to CMap and GBrowse • Blast results are imported into CMap and GBrowse with additional information such as trace files and FPCs
Minimum Tile Path Pipeline Usage • A table of alignments between the seed BAC and the BAC end sequences contains links to CMap and GBrowse. • CMap displays the FPC data for the seed BAC and the potential next BACs. • GBrowse provides an alignment of the BES with the seed sequence and displays the trace data.
Maize Production Sequencing • Shotgun of 19,000 BACs • Fosmid End Sequencing of 1 Million Reads • BAC End Sequencing of 220,000 clones
Maize BAC shotgun BAC DNA received from AGI or prepared at the GSC Small Scale Library Construction Production Sequencing - 1,536 reads/project Automated Shotgun_done
www.maizesequence.org Sequenced BAC FPC Contig Virtual Bin Core Bin Marker Chromosome Synteny Views Main Navigation bar is accessible from every page Contains multiple entry points to the genome
MapView Displays statistics by chromosome and provides entry points based on a single chromosome
CytoView Provides detail information on features anchored to the FPC map. The side bar highlights the location on the chromosome and provides page specific functionality including data export. The Detailed view is customizable, tracks can be added or removed by the users. Feature contain drop down menus that contain general information as well as provided internal links, and external links.
ContigView This view is based BAC coordinated and displays annotation levels II and III. The header contains the Clone name in the physical map, GenBank Accession, and Chromosome and FPC contig information. Detailed view offers semantic zooming, customizable and provides links to other views and information resources.
“Methyl-filtering” Approach for Genome Sequencing Discard methylated DNA gene hyper-methylated region Probe BAC library with unmethylated DNA unmethylated (gene-rich) region Sequenced BACs © Orion genomics
Repeated sequence Gene sequence Enrichment for maize genes using methylation filtration Relative amount Maize DNA unfiltered Maize DNA filtered
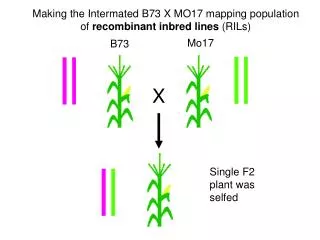
Maize Missouri 17 “chromosome 10” project update Dan Rokhsar 3 October 2006
Aims: “Plan A” • Generate and annotate “gene space” for the ~180 Mbp chromosome 10 of Mo17 using a random shotgun approach from flow-sorted chromosomes. • This resource will complement the BAC-by-BAC sequencing of B73, informing our understanding of intra-species variation, from SNPs to chromosomal organization. • The project will serve as a pilot R&D study for chromosome-scale random shotgun sequencing of complex genomes
Challenges • Produce high-quality shotgun library from a single chromosome (year 1) • Apply flow sorting methods to root tip preparations or oat-maize hybrid lines with maize Mo17-10 • Assemble shotgun sequences and relevant mapping data to recover non-repetitive and ‘distinguishable repetitive’ regions (years 1-2) • DuPont Mo17 BAC library, BAC-end sequence • Targeted mapping to link across complex repeats • Targeted finishing of “gene space” from whole-chromosome-shotgun draft (year 2) • Interplay of finishing with annotation
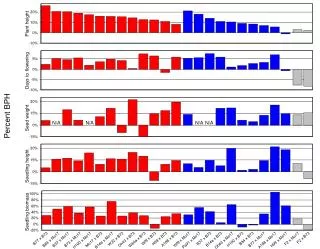
Project goals for researchers and breeders • Unlimited markers for mapping • Nearly complete gene set for Mo17-10 • Conserved synteny/chromosome dynamics with sorghum • Evolutionary approaches empowered • Novel reagents begin to emerge • Framework for understanding strain differences
Milestones • Year 1 • Produce test libraries from mock flow sorted material (JGI) • Produce preliminary flow sorting data for discussion at Advisory Committee meetings (NFCR) • Produce 1-10 micrograms of flow sorted chromosome 10 material (NFCR). • Complete library production (JGI) • Begin shotgun sequencing, with associated data deposition (JGI)
Milestones • Year 2. • Complete initial shotgun assembly, with associated data deposition (JGI) • Integrate with physical map data from DuPont (JGI) • Complete two rounds of primer walking (SHGC) • Annotate initial draft assembly, with data release (JGI) • Complete subsequent rounds of targeted finishing reactions (SHGC) • Complete physical mapping of markers and release to public repositories (PGML) • Produce final assembly incorporating finishing data (JGI, SHGC) • Publish detailed analyses of Maize Genome Project outcomes (all) • Offer summer course on maize genome data (JGI)
Even in expert hands, purity of chromosome prep is 85-90% • Li, Arumuganathan, et al. Flow cytometric sorting of maize chromosome 9 from an oat-maize chromosome addition line. TAG (2001).
Alignment of Mo17 “gene space” with B73 allele ~97% identity • In unique “genic” regions (especially coding sequence), can easily align Mo17 and B73 to detect polymorphism. • Cf comparable human-chimp alignments at ~98.5% • (putative aminotransferase, Morgante et al.) • Mo17 1 AACCAATTGGCAGCATTATTATTTTGAACAGATAAAAATCACGCCAGGGCGATGGATACT 60 • B73 88023 ..............C.........C................................... 88082 • Query 61 CAGCTCAATCACGGAATTCATCCATGAACTTCTCGTGGAACTCCTTGAGCCTGGATACTA 120 • Sbjct 88083 ............................................................ 88142 • Query 121 TCGCAGGTATCTTGTCCTCCTGCGGCAGTATCGTGCACCTGAAGTGCCACGTTCCAGGGA 180 • Sbjct 88143 ............................................................ 88202 • Query 181 CCTTCA--------CG--G-T--G-T-C-GC-AAAGCAACGTGTCAGTATCGTGTGCATC 223 • Sbjct 88203 ......CGGTGTCG..AA.T.AA.A.C.A..A................G........... 88262 • Query 224 TGAAGCTTAACGATGCTTTGAAACGGCAGGGACTTCCACaaaaaaaGG-CTTTTGAGATT 282 • Sbjct 88263 .............................................G..G........... 88322 • Query 283 ACCCACCTGTCCAAACCCAGAACCGGGGACGACGACGATTCCAGTGGCTTCCAGTAGGCG 342 • Sbjct 88323 ............................................................ 88382 • Query 343 TTTTGCGTAGTATGCATCTGGCGCAGTGCCGACTGCTTGGGCAGCTCCAATTGCCTTCTG 402 • Sbjct 88383 ..........................................T................. 88442 • Query 403 GGGTAAATGAAGGCGTGGGAACAGATACATTGCACCTTCGGCTTTGTTGCATGTAATTCC 462 • Sbjct 88443 ............................................................ 88502 • Query 463 TTCTAAACTGTTGAATGCTTCTTCCAAAGCCTGTGACAGAAGAACACGTAACAATAAGAA 522 • Sbjct 88503 ............................................................ 88562 • Query 523 GGTGCTTATAAGATTCAGGaaaaaaaa--TCTTTTTTAAAGTTGTTTTGCATATGTTAAC 580 • Sbjct 88563 ...........................GA............................... 88622 • Query 581 GGACTACTCGACCAGGGGTATAGCTTTTATTCTTGTTTGATATTTCCATATTAGGACTCT 640 • Sbjct 88623 ..........G................................................. 88682
JGI Sorghum update • Sorghum WGS currently at ~7X (in Trace Archive) • mostly small insert plasmids sequenced to date • BAC-end and fosmid-end sequences coming by end 2006 • but uniformity of BAC library is in question, may limit assembly • Quick and dirty assemblies look good using “skeleton” of method proposed for maize • ~13 kb contigs and ~300 kb scaffolds (N50 #’s) at ~5X • considerable scaffolding even without much BAC/fosmid data • recovering ~2/3 of genome is easy even setting aside “difficult” repeats, as predicted for maize • Expect full 8X assembly (with map integration) ready late Q1 2007. • Quick and dirty annotation: ~42,000 genes in low copy families • plus >100K retrotransposon-ish genes even in easy-to-assemble regions
Early peek at Sorghum-rice comparison shows syntenic segments Sorghum-Rice syntenic segments are of uniform molecular “age” Comparable to human-chicken divergence Younger than Rice-Rice paralogs (from cereal-specific duplication) Transversions/synonymous site Loci in syntenic block
Maize divergences (transversions) Maize: 7,960 complete/29,922 partial peptides Sorghum: 5,927 complete/19,681 peptides Sugarcane: 6,566 complete/ 21,850 peptides ~16,000 gene families at base of grasses ~12,000 families defined by rice/arabidopsis/poplar sugarcane sorghum Arabidopsis rice
Gene Ontology (GO) Annotation • EST and Methyl Filtered Assemblies • BLAST vs Uniprot • Database of Uniprot-to-GO associations queried • GO numbers categorized and tallied
Biological Process Molecular Function EST MF EST Cellular Component MF EST MF EST and Methyl Filtered GO Classifications EST data is displayed on inner ring Methyl Filtered data on outer ring
Kyoto Encyclopedia of Genes and Genomes (KEGG) • Collection of databases provide visual representation of enzymatic pathways • Can be used to identify which subset of a pathway has been identified in a given organism • Provides an additional tool for comparative genomics
Obtaining KEGG Diagrams • Methyl Filtered Assembled Data • BLAST vs Uniprot • Uniprot results translated to EC numbers • EC numbers uploaded to KEGG
Color Key: Tobacco Alone Solanaceae Alone Tobacco + Solanaceae Tobacco vs. Solanaceae Unigenes
Gene Annotation Data Repository • We are compiling all current gene data into a searchable database with a web-based user interface. • We are adding computational annotations • Blast • FGenesH • Glimmer • UniProt • EST Homology • Human Manual Curation • Manual assessment of all computational annotation to assign an annotation • The Web interface will show strength of annotation and provide links to supporting data.
Old Pipeline vs. New Pipeline • Change in High Quality base pairs Increased Decreased Methylfiltration 125,745,207 Non-methylfiltration 3,450,307 ESTs 132,543 Sheared BACs 11,789,937 BACends -55,418 Total base pair increase: 141,056,576 Percentage bp increase 17.9%
Post Process Data Comparison • Analysis of differences in data run through our Old Pipeline and our workflow based method for identical trace files. • Sequences are the total number of sequences used for this/ bases are the total number of bases for those sequences. • Longest sequence: longest sequence in base pairs, post processing • Shortest sequence: shortest sequence in base pairs, post processing • Average sequence length: average number of bases per sequence. • Difference: is the change in the number of base pairs processed in the Old Pipeline compared with the workflow based method. (number for the workflow method minus the number for the Old Pipeline row). • Increase in Bases: is the percent increase in information provided by the workflow method as compared to our old pipeline.
Search tool • Key Word search • Selectable databases • Output shows Which database hit occurs in and strength of annotation for the given key word hits
Introducing Tobacco and our Approach to Sequencing • Analysis of EST sequencing • Sequencing by Methylation Filtration • Targeted BAC sequencing • Functional Analysis and Annotation • Conclusions