Download

1 / 22
250 likes | 588 Vues
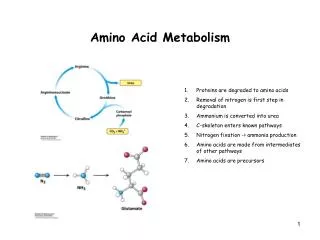
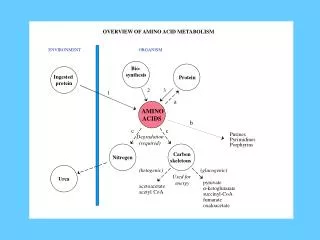
Amino acid metabolism V. Enzymopathies related to amino acid metabolism. Figures : Lehninger-4ed; chapter: 18 (Stryer-5ed; chapter: 23). I. Urea Cycle Defects (UCD’s). • Lack or defect of urea cycle (or any of its enzymes) is crucial for life

E N D
Amino acid metabolism V.Enzymopathies related to amino acid metabolism Figures: Lehninger-4ed; chapter: 18 (Stryer-5ed; chapter: 23)
I. Urea Cycle Defects (UCD’s) •Lack or defect of urea cycle (or any of its enzymes) is crucial for life •A typical result of UCD’s (except arginase defect) is hyperammonemia or the build-up of one or more urea cycle intermediates (depending on the missing enzyme) • Permanent activation of glutamate dehydrogenase also cause hyperammonemia (hyperinsulinism-hyperammonemia syndrome)
Possible treatments: • low protein diet (strict dietary control and supplements of essential amino acids) • removal of excess ammonia • refill of urea cycle intermediate pools • careful administration of aromatic acids (benzoate and phenylbutyrate) in the diet can help lower the level of NH3 in the blood
• Gly and Gln used up must be regenarated in reactions that take up NH3 •hippurate and phenylacetylglutamine are nontoxic and are excreted in the urine • this pathways become prominent when aromatic acids are ingested
N-acetylglutamate synthase deficiency: • results in the absence of N-acetylglutamate (normal activator of carbamoyl phosphate synthetase I) •treatment: administering carbamoyl glutamate (activator of carbamoyl-P synthetase I)
Deficiencies ofornithine transcarbamoylase, argininosuccinase, argininosuccinate synthetase are treated by supplementing the diet with Arg! In arginase deficiency (rare) Arg excluded from diet
Neurotoxic effects of hyperammonemia • Hepatogenic encephalopathy • Ammonia easily crosses blood-brain barrier • It is annihilated or scavenged in glutamate dehydrogenase reaction, while consuming -ketoglutarate • Abnormal depletion of -ketoglutarate decreases the rate of TCA cycle, in an extreme case to 0 energy production slows down or may even stop • (Excess ammonia consumes glutamate, a precursor of GABA - an important neurotransmitter - in the glutaminase reaction)
II. Genetic disorders of the amino acid degradation 1.) Nonketotic hyperglycinemia ●Defect of the glycine cleavage enzyme (Gly degradation) ●Elevated serum levels of Gly severe mental deficiencies and death in very early childhood (Gly is an inhibitory neurotransmitter, perhaps explaining the neurological effects of the disease.)
2.) Methylmalonic aciduria • Defect of methylmalonylCoA isomerase (methylmalonylCoA succinyl-CoA) • Ketoacidosis, mental retardation, early death • Treatment:Vitamin B12, administration of controlled amounts of the amino acids involved
Defect of the branched-chain -keto acid dehydrogenase complex (degradation of Leu, Ile and Val) • Lethal in days after birth (vomit, spleen) if not, it causes mental retardation • Urine has a characteristic odor after day 6-7 (the -keto acids accumulate in the blood urine) • Treatment:Administration of a diet with strictly controlled amounts of Leu, Ile, Val
4.) Homocystinuria I. • Defect of cystathionine -synthase (Met degradation) • Mental retardation, thrombosis in arteries and veins • Treatment: Vitamin B6, diet rich in Cys and poor in Met
5.) Histidinaemia • Defect of histidase or histidine-amino lyase (His degradation) • Mental retardation (causal relationship not proven yet) • Treatment:Controlled administration of His
Genetic disorders of phenylalanine degradation6.) Phenylketonuria (PKU) • Defect of phenylalanine hydroxylase (Phe degradation) • Mental retardation(phenylpyruvate inhibits pyruvate decarboxylase in the brain and the formation of myelin; it has influence on the levels of different neurotransmitters as well)Inhibits Trp metabolism as well • Treatment:Diet poor in Phe and Tyr (only for protein synthesis!)
Defect of dihydrobiopterin reductase can also cause PKU! Tetrahydrobiopterin is required for the formation of L-dopa and 5-hydroxy- tryptophan (precursors of norepinephrin and serotonin) In this type of PKU, these precursors must be supplied in the diet!
Alternative pathway for catabolism of Phe in PKU: Phenylpyruvate, phenylacetate and phenyllactate can accumulate in tissues, blood, and urine. The characteristic odor of the urine is due to the phenylacetate.
7.) Tyrosinaemia II (Richner-Hanhart Syndrom) • Defect of cytosolic (soluble) tyrosine aminotransferase (Phe/Tyr degradation) • Ulcers, keratosis, keratitis, mental retardation, p-hydroxy phenyllactate accum. in urine • Treatment: Diet poor in Phe and Tyr
8.) Tyrosinaemia III • Defect of para hydroxyphenyl pyruvate dioxygenase (Phe/Tyr degradation) • Mild mental retardation, drowsiness, ataxia
9.) Alkaptonuria • Defect of homogentisate dioxygenase (Phe/Tyr degradation) • Urine darkens on standing (black), arthritis •Treatment:Ascorbic acid diet poor in proteins
10.)Tyrosinaemia I (tyrosinosis) • Defect of fumarylaceto- acetase (Phe, Tyr degradation) • Hepatic cyrrhosis, dilatation of microtubules in the kidney, urine with characteristic odor •Treatment: diet poor in Phe and Tyr
11.) Albinism • Defect of the tyrosine 3-monooxygenase (tyrosinase) (melanine synthesis from tyrosine) • lack of pigmentation: white hair pink skin