BASIC PHARMACOKINETICS
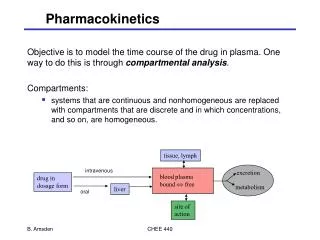
BASIC PHARMACOKINETICS. FATE OF DRUGS IN BODY. DRUG ADMINISTERED. (ABSORPED). INTENDED EFFECTS. METABOLISED. ENTER SYSTEMIC CIRCULATION. ELIMINATED. REACH SITE OF ACTION. DISTRIBUTED. Pharmacokinetics and pharmacodynamics.


BASIC PHARMACOKINETICS
E N D
Presentation Transcript
FATE OF DRUGS IN BODY DRUG ADMINISTERED (ABSORPED) INTENDED EFFECTS METABOLISED ENTER SYSTEMIC CIRCULATION ELIMINATED REACH SITE OF ACTION DISTRIBUTED
Pharmacokinetics and pharmacodynamics • Pharmacokineticsis the study of how a drug reaches its target in the body and how it is affected on that journey, i.e; effect of the body on the drug. • the study of how is the drug absorbed, distributed, metabolized and excreted in the body • Pharmacodynamics is the study of how drugs interact with a molecular target, i.e; effect of the drug on the body.
ADMINISTRATION • Aim of drugs administration is to achieve therapeutic level at site of action. • Route of drugs administration : First category – drugs must pass through a barrier to reach systemic circulation. 1. Oral 2. Inhalational 3. Injection into the body but not the vascular system (IM,SC) 4. Through natural orifices (rectal, nasal & vaginal)
ADMINISTRATION Second category – drugs injected directly into the systemic circulation (IV) Third category – Topicals through skin (stratum corneum) Fourth category – Drug administered by these specialized routes of administration achieve local and targeted therapy. A portion may reach systemic circulations (SA. Epidural, intraocular)
ADMINISTRATION 1. Oral Administration • Most convenient, most economical • Disadvantages: • emesis (drug irritation of the gastrointestinal mucosa) • digestive enzymes/gastric acidity destroys the drug • unreliable or inconsistent absorption due to food or other drug effects • metabolism of the drug by gastrointestinal flora
Factors determining rate of drug effect onset 1. Dissolution 2. Absorption 3. Significant first pass effects 4. The mucosal surface area 5. Gastric emptying 6. Perfusion to the gut mucosa
2. Transdermal Administration The delivery of drugs through an intact skin. The stratum corneum is the main barrier to the diffusion of drugs, especially polar compounds. Potent drugs with high lipid solubility can be absorbed transdermally to produce systemic effects (e.g. fentanyl, GTN, ethinylestradiol, hyoscine). The stratum corneum will also act as a reservoir for lipid soluble drugs for several days after the drug has been stopped. ADMINISTRATION
Advantages: • sustained, therapeutic plasma levels (reduced peaks/valleys associated with intermittent drug administrations) • avoids continuous infusion technique difficulties • low side effect incidence (smaller doses)
ADMINISTRATION • Factors contributing to reliable transdermal drug absorption: • molecular weight < 1000 • pH range 5-9 in aqueous medium • no histamine-releasing action • daily drug requirement <10 mg • Example of drugs available for transdermal delivery: fentanyl, GTN
ADMINISTRATION 3. Rectal Administration • Proximal rectum administration Absorption into superior hemorrhoidal veins then enters the portal venous system then to the liver (possible first pass hepatic effect) and finally into the systemic circulation • Low rectal administration of drug may allow the drug to enter the systemic circulation without passing through the liver • Generally unpredictable pharmacological responses for the above reasons • Rectal mucosal irritation possible
ADMINISTRATION 4. INTRAMUSCULAR • Less painful than subcutaneous injection • Muscle less prone to chemical injury and infection than SC fat • Irritating substances may be given • Uptake of drugs following IM injection is more rapid, especially for water-soluble drugs • Duration of action shorter following IM than SC injection. • Absorption is dependent on perfusion • Unpredictable in the very emaciated and obese • Complications – Sterile abscess, Sciatic nerve injury, Pain
ADMINISTRATION 4. SUBCUTANEOUS • Advantages – absorption is slow and sustained • Improve by i) hyaluronidase ii) implanting insoluble pellets – contraceptive iii) dissolving in oil – penicillin • Time of onset – 30 mins, peak – 60 mins • Skin blood flow – 12.8 ml/100g/min • Decreases absorption when vasoconstricted
ADMINISTRATION 5. INTRAVENOUS • No absorption required, bypass liver • Accurate and predictable • Fast: able to titrate in ill and old patient • Irritating substance may be used (endothelium relatively insensitive) Disadvantages - No retreat - IV access required - Unfavourable reactions secondary to high plasma concentration
ABSORPTION Absorption : passage of a drug from an external site of administration across body tissues to the systemic circulation / site of action. Absorption occurs through : 1. passive diffusion 2. special carrier mechanism 3. pinocytosis
Pinocytosis is theingestion of dissolved materials by endocytosis. The cytoplasmic membrane invaginates and pinches off placing small droplets of fluid in a pinocytic vesicle. The liquid contents of the vesicle is then slowly transferred to the cytosol.
ABSORPTION • Absorption occurs mainly by passive non-ionic diffusion and is therefore dependent on factors such as 1. Lipid solubility 2. Molecular size 3. Ionisation*
ABSORPTION • Fick’s law of diffusion • Rate of diffusion A x D x (P1-P2) T • A = surface area • D = diffusion coefficient = solubility/ MW • T = thickness • P1-P2 = concentration gradient
IONIZATION • Drug ionization reduces a drug's ability to cross a lipid bilayer • Amount of ionization depends on pKa (dissociation constant) • Numerically equal to the pH of a solution in which the drug is 50% ionised • pKa of a drug is the measure of the acid strength, e.g. an acid with a pKa of 2 is 10X as strong as an acid with pKa of 3 • The basic strength of drugs increases with the pKa, a basic drug of pKa of 9 is 10X as strong as base with pKa of 8.
The equilibrium constant K is defined as the fraction of H+ and A- to the HA. K = ([H+] [A-] ) / [HA] for this equilibrium K is called Ka for acid dissociation constant. Since we are interested in the amount of H+ we need to arrange the equation to just have [H+] in one side. [H+] = Ka ([HA]/[A-] ) Taking the logarithm of each side gives: log [H+] = log Ka + log ([HA]/[A-] ) Now we can multiple by -1. -log [H+] = -log Ka - log ([HA]/[A-] ) Which is the same as: -log [H+] = -log Ka + log ([A-] /[HA]) Traditionally, -log is substituted with a lower case p, so the equation becomes (known as the Henderson-Hasselback equation: pH = pKa + log ([A-] /[HA])
So if we know the pH of a solution and the pKa we can determine how much [A-] and [HA] we have in solution, using the rearranged equation below. [A-] /[HA] = exp (pH - pKa) This means that when pH is equal to pKa, [A-] = [HA], or that the acid is 50% ionized.
Ionization - Weak Acids • eg Aspirin (pKa 3.5) • RCOOH RCOO- + H+ • pH = pKa + log [ionized] [unionized] • Weak acid; • >acidic; > unionized • >basic; >ionized
Ionization - Weak Bases • eg Ephidrine (pKa 9.6) • RNH3+ RNH2 + H+ • pH = pKa + log [unionized] [ionized] • Weak base; • >basic; >unionized • >acidic; >ionized
ION TRAPPING • A phenomenon whereby drugs are kept in environments of altered pH, by the change in equilibrium brought about by the change in pH
Placental Transfer Of Basic Drugs • e.g local anesthetics • fetal pH is lower than maternal pH • lipid-soluble, nonionized local anesthetic crosses the placenta converted to poorly lipid soluble ionized drug • gradient is maintained for continual transfer of local anesthetic from maternal circulation to fetal circulation • in fetal distress, acidosis contributes to local anesthetic accumulation
Kidney - Nearly all drugs filtered at the glomerulus - Most drugs in a lipid-soluble form will be reabsorbed by passive diffusion. - To increase excretion: change the urinary pH to favor the charged form of the drug since charged form cannot be readily reabsorbed (they cannot readily pass through biological membranes) • Weak acids: excreted faster in alkaline pH (anion form favored) • Weak bases: excreted faster in acidic pH (cation form favored)
Bioavailability • The proportion of intact/unchanged drug that is available to the systemic circulation, after oral/intramuscular administration. • Affected by: • Extent of absorption • First Pass metabolism
IV PO Bioavailability = AUCPO X 100% Quantifies Absorption AUCIV
First Pass Metabolism • Proportion of drug that is removed or metabolised by an organ before it reaches the systemic circulation during the initial single transit • Liver • High ER (>0.7) eg Propanolol, Lignocaine • Low ER (<0.3) eg Warfarin, Thiopentone • Gastrointestinal eg; L-dopa, Chlorpromazine • Pulmonary eg; Lignocaine, Propanolol, opiods
DISTRIBUTION • After gaining access to the systemic circulation, drugs distribute in different tissues and organs of the body. • Factors affecting; • Physicochemical char. of the drugs • Lipid solubility • Degree of ionization • Cardiac output & regional blood flow to various organs: VRG, MG, FG • Binding: Plasma protein / Tissues
Volume of Distribution • Quantifies distribution. • Volume of distribution (Vd) of a drug is a mathematical expression of the sum of the apparent volumes of the compartments that constitute the compartmental model. • This value depicts the distribution characteristics of a drug in the body. • The volume in which the amount of drug in the body would need to be uniformly distributed to produce the observed plasma concentration.
Volume of distribution is calculated as the dose of drug administered intravenously divided by the resulting plasma concentration of drug before elimination starts. Vd = Amount of Drug in the Body Plasma Drug Concentration • Indicates the extent of extravascular tissue uptake of the drugs
Volume of Distribution Factors determining Vd 1. Physicochemical characteristics of the drug • Lipid solubility of drug • Degree of ionisation (function of drug pKa and ambient pH) • Molecular weight 2. Degree of plasma protein binding (acidic and neutral drugs to albumin and basic drugs to 1-acid glycoprotein) 3. Tissue binding 4. Regional blood flow to tissues (affected by age and disease states eg CRF, CCF)
Volume of Distribution Clinical Significants Drugs with small Vd (5-20 litres) are predominantly localized in plasma/ECF (muscle relaxants) or are extensively bound to plasma proteins (warfarin, phenytoin, tolbutamide). These are typically highly polar, water-soluble drugs. Drugs with Vd equal to TBW (30-45 L in adults) are evenly distributed throughout TBW (examples are alcohol, urea and some sulphonamides) Drugs with Vd larger than TBW have extensive tissue penetration and binding to tissues or sequestration in fats. In these circumstances, the concentration in tissues may be higher than in plasma. These are typically highly lipid soluble drugs for example digoxin, fentanyl, thiopentone, most phenothiazines a most anti-depressants.
Protein Binding • Drugs are bound to plasma protein in a reversible manner • Saturable process • 3 main types of carrier proteins for drugs: 1. Albumin – bound mainly to acidic drugs e.g diazepam, digoxin, warfarin, barbiturates 2. α1-acid glycoprotein – bound mainly to basic drugs e.g. opioids, β-Blockers, Tricyclic anti- depressants, LA, antiarrhythmics 3. Globulins – Steroid hormones, Thyroid hormone
Protein Binding • fraction unbound (fU) = unbound [drug] total [drug] • fU is determined by i) The affinity of the drug for protein ii) The concentration of the binding protein iii) The concentration of the drug relative to that of binding protein
Protein Binding Implication 1. Distribution 2. Clearance - bound and free drug can be cleared by liver and kidney - in liver, ↑ ER (> 0.7) insensitive to protein binding but for ↓ ER (<0.3), dependent on [free drug] - in kidney, glomerular filtration dependent on protein binding 3. Disease Process - altered in renal or liver diseases 4. Drug interaction
METABOLISM • Role of metabolism or biotransformation is to convert pharmacologically active, lipid-soluble drugs into water soluble and often pharmacologically inactive metabolites. • Increased water solubility decreases the Vd and enhance its excretion. • Main site liver, others are kidney, lung, plasma • Drug biotransformation mechanisms are described as either phase I or phase II reaction types.
Phase I Metabolism (non synthetic reactions) • Usually result in drug oxidation, reduction or hydrolysis. • Most phase 1 reactions are carried out by the liver cell in the smooth ER or microsomes • A non-specific enzyme system in the ER (Cytochrome P-450 or mixed function oxidase system) is responsible for most drug oxidations and reductions and for some hydrolytic reactions
Cytochrome 450 System Gain of electron (e-) result in reduction Loss of e- results in oxidation oxidation-reduction process Involves two important microsomal enzymes 2. Flavoprotein • Cytochrome P450 Induce Inhibited Inactivated
Phase II metabolism (synthetic or conjugation reactions) • Combinaton of unchanged drugs or phase I metabolites with other chemical group (eg; glucuronide, sulphate, acetate etc) • The most important reaction is conjugation of drugs to glucuronides • Conjugates are often polar and inactive • Occurs both in microsomes or cytosol