Download

1 / 28

E N D
Medical University of VarnaPostgraduate Programme in Internal Medicine“Pathophysiology of Cardiovascular System”PATHOPHYSIOLOGY OF HYPERTENSIONApostolos I. HatzitoliosAssistant Professor of Internal Medicine1st Propedeutic Medical Department Aristotle University of ThessalonikiDepartment of Hypertension and Vascular DiseaseAHEPA Hospital
Pathophysiology of Arterial Hypertension Structural hypertrophy of the heart and vessels sustains hypertension and creates a vicious circle of further increase through kidney damage
MECHANISMS OF HYPERTENSION CARDIAC OUTPUT/PERIPHERAL VESSEL RESISTANCE CARDIAC HYPERTROPHY Although considered compensatory mechanism to an increased vascular resistance ,it could also reflect a primary response to repeated neural stimulation and thereby could be an initiating mechanism of hypertension by increasing cardiac output. INCREASED FLUID VOLUME Increased preload could induce hypertension by increasing cardiac output in pre- and mild hypertension, since in most studies subjects with high blood pressure (increased peripheral vessel resistance) have lower blood volume (and total exchangeable sodium).
RELATION OF BLOOD VOLUME TO BLOOD PRESSURE The redistribution of blood volume because of peripheral vessel constriction causes increase in venous come-back, cardiopneumonal circulation and cardiac output. AUTOREGULATION MODEL The initial high cardiac output gives way to increased peripheral vessel resistance (intrinsic property of the vascular bed to regulate the blood flow depending on the metabolic need of tissues through constriction and structural thickening ). Juliusproposes an other model: structural changes decrease the cardiac responses to nervous and hormonal stimuli but enhance the vascular responses causing vascular hypertrophy and altering the wall-to-lumen ratio.
EXCESS SODIUM INTAKE Epidemiologic evidence Primitive people who do not eat sodium have no hypertension.In groups of people with the same way of life hypertension depends on sodium intake. Experimental evidence When hypertensives are sodium-restricted their BP falls. Increased NaCl intake increases BP by activating mechanisms like increasing intravascular volume and intracellular sodium and calcium, causing vasoconstriction, insulin resistance, and increased catecholamines . Sensitivity to Sodium Although increased sodium intake is the common issue in industrialized societies, the fact that only 20-50% develop hypertension suggests a variable degree of BP sensitivity to sodium ( that is increased reabsorption in renal tubules), in which both heredity and interaction with environmental exposures are involved.
Mechanisms interpreting sensitivity to sodium • Increased activity of the sodium-hydrogen exchanger in the proximal tubule • Increased sodium reabsorption because of decreased renin-aldosterone suppressed production • Increased calcium input in vessel smooth muscles • Increased sympathetic nervous system activity • Endothelial dysfunction related to decreased nitric oxide response to sodium loads • Inherited, especially maternal (polymorphism 2 of ACE gene) • Increased Angiotensin II and decreased bradykinin receptors.
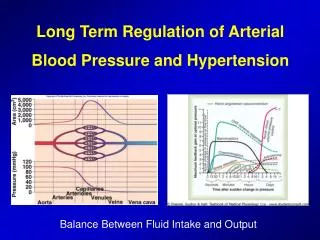
RENAL SODIUM RETENTION Apart from sodium sensitivity, for BP development a possible defect in the pressure-natriuresis mechanism could be responsible, which blocks BP return to normal ( Guyton 1992 ) , by affecting natriuresis curve (through preglomerular vasoconstriction and increase sodium reabsorption in the tubules). Ouabain (endogenous glycoside ) , peptide produced in adrenal gland cortex, could be responsible for hypertension with increased blood flow. Apart from that, since ouabain is an inhibitor of Na-K ATPase pump causes increase of intracellular sodium and calcium in vessel smooth muscles, increasing thus the resistance. It is also mentioned from experimental data that ouabain causes hypertension through increased endothelin production.
NATRIURESIS CURVE: ITS SHIFT TO THE WRIGHT IS A MECHANISM CAUSING HYPERTENSION.
NEPHRON HETEROGENEITY Sealeytheory: a subpopulation that is ischemic from either afferent arteriolar vasoconstriction or from an intrinsic narrowing of the lumen causes increased renin secretion from this population . This renin secretion interferes with the compensatory capacity of normal nephrons to adaptively excrete sodium and ,consequently, perturbs overall blood pressure homeostasis. REDUCED NEPHRON NUMBER Breneret al in1988υproposed that hypertension may arise from congenital reduction in the number of nephrons limiting the ability to excrete sodium raising BP and setting of a vicious circle whereby systemic hypertension causes glomerular hypertension. These investigators point out that as many as 40% of individuals younger than 30 years have fewer than 600.000nephrons/kidney and speculate that those individuals whose congenital nephron number fall in the lower range are susceptible to the development of essential hypertension.
LOW BIRTH WEIGHT Large studies showed that BP in adulthood is reversely analogous to birth weight possibly due to congenital oligonephropathy (less nephrons) because of intrauterine growth retardation. Causes of low birth weight. Increased number of pregnancies in adolescence Small intervals between pregnancies Inadequate nutrition
angiotensinogen renin kidneys negative feedback angiotensin I Increased BP bradykinin ACE(kininase) inactive peptides Sodium retention angiotensin II receptors ΑΤ1,ΑΤ2 aldosterone AT1 Vessel receptors AT1 receptors in adrenal glands VASOCONSTRICTION RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM (RAAS) Tanin Cathepsin G Tissue plasminogen activator Alternative ways reverse AT1 action
RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM (RAAS) • The main mechanism throughRAAScontributes to hypertension is a negative feedback in the axon renin-angiotensin-aldosterone in kidneys and adrenal glands. Furthermore angiotensin II causes reduced production and action of nitric oxide by altering arachidonic acid metabolism, thus increasing vasoconstrictive prostaglandines and increased endothelin production. • Although low renin is expected in essential hypertension, most patients have normal or even high rates. • Possible mechanisms: • Nephron heterogeneity • Defect in negative feedback • Increased SNS activity • IN hypertensive patients with high renin activity, the main mechanism is vasoconstriction due to increased angiotensin II. • In hypertensive patients with normal renin activity, the main mechanism is increased intravascular volume.
HYPERTENSION AND NERVOUS SYSTEM • Increased SNS activity in central nervous system (SNS and hypothalamus) and peripheral nervous system (adrenergic ending, α1, α2 και β-vessel receptors) . • Contribution of SNS in hypertension development through interaction with renin-angiotensin-aldosteron axis. • S tress causes SNS activation and increased sodium retain, insulin resistance ( increased tissues metabolism) • Baroreceptor malfunction: Normally, baroreceptors are activated from increased BP and central venous pressure, leading to vagus stimulation, SNS inhibition and BP decrease. In hypertensives this respond decreases because of structural and functional receptor changes. • Stress reaction : Normotensives with high risk for hypertension development show a greater SNS and cardiovascular stress respond. Production of vasodilators is reduced and smooth muscle multiplication is increased.
PERIPHERAL RESISTANCE (PR) • Hypertension is maintained by increased PR due to decreased arterial lumen size or radius in small resistance arteries or arterioles. According to Poiseuille’s law vascular resistance is inversely related to the radius of the fourth power (decreased lumen size in vasocontraction or hypertrophy→increased PR) • In studies of small resistance vessels from subcutaneous tissue of hypertensive subjects compares to normotensives, increases in the ratio of media thickness to internal diameter of 26-62% have been recorded. • Mechanisms of vessel remodeling • Remodeling of smooth muscles around the smaller lumen size. • In increased BP (e.g. increased angiotensin II) vessel hypertrophy is caused due to genetically inducedhypertensive reaction of the vessels in growth factors ( PDGF-A , HGF ) .
CELL MEMBRANE ALTERATIONS Να-Η Countertransport Increased countertransport in hypertensives, causing increased vascular tone, cell growth (Na, Ca entry), left ventricular hypertrophy and increase Na reabsorption in renal proximal tubuls ( possibly related to a polymorphism of a-adukine gene and change in tropomyocine expression) Alterations in cell membrane structure Increased cholesterol/phospholipid ratio leading to increased Na permeability (increased Να-Η pump activity) Pathologic Na and Ca transport intracellularly, causes increase PR.
Hypothesis linking abnormal ionic fluxes to increased peripheral resistance.
OBESITY AND HYPERTENSION • Obesity causes hypertension through: • Increased cardiac output and volume expansion • Increased Leptin, causing : • Increased SNS activity • Increased NO decomposition • Increased Na reabsorption • Shift to the right of the natriuresis curve • Increased renin activity • Fatty infiltration of the kidney causing increased glomerular pressure and thus their damage • Increased insulin resistance and hyperinsulinemia
MECHANISMS THROUGH WHICH INCREASED INSULIN RESISTANCE AND HYPERINSOULINEMIA CAUSE HYPERTENSION • MECHANISM n • Increased sodium-aqua reabsorption (Gupta et al 1992) • Increased sensitivity in sodium consumption (Sharma et al 1993) • Increased response of BP and aldosterone to angiotensin II (Rocchini et al 1990) • Alterations in transmembran ion exchange nn • Increase of intracellular sodium (Barbagallo et al 1993) • Decreased Na/K ATPase activity (Pontremoli et al 1991) • Increased Na/H pump activity (Aviv 1992) • Increased intracellular calcium (Aviv 1992) • Growth factors activation especially in vessel smooth muscles (Bornfelt et al 1992) • Increased SNS activity (Lembo et al 1992) • Decreased vasodilator prostaglandines (Axelrod 1991) • Impaired vasodilation (Baron et al 1993) • Increased endothelin production (Hu et al 1993)
CENTRAL ADIPOSITY HYPERINSULINE-MIA IGT CORONARY DISEASE INSULIN RESISTANCE SYNDROME HYPERTENS DYSLIPIDE-MIA MICRO- ALBUMINOURIA HYPERURICE-MIA METABOLIC SYNDROME Χ
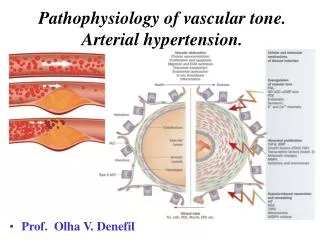
VASOACTIVE FACTORS –REGULATION OF VESSEL TONE Vasoconstrictive: TXA2, PGH2, O2, A II Vasodilative: NO, GGI2 ,EDHF Blood vessel Growth inhibitors: NO Growth inductors: Endothelin- 1, TXA2,O2,A II
ENDOTHELIAl DYSFUNCTION (surface of 400m2, 1.200.000.000 cells:the largest endocrine gland) Decreased synthesis (endothelial synthase, e-NOsacts in the basic aminoacid L-arginine),possibly through decreased response to acetylcholine ( dilation through muscarine receptors in intact endothelial cells , contraction through muscarine receptors of smooth muscle cells in damaged endothelium) or increased NO decomposition or/and increased endothelin production NO reaches smooth muscle cells, stimulates guanylcyclase and production of 3,5 cyclic monophosphoric guanocin (cGMP). Ach, bradykinin, P substance and others, by acting in receptors on endothelial cells increase Ca++ entry and activate e-NOs. NO apart from vasodilation inhibits smooth muscle cells’ multiplication (hypertrophy) and ET-1 production. NO system in essential hypertension pathogenesis Decreased NO production under normal circumstances and stimulation Decreased vasodilation as response to Ach Increased vascular resistance Essential hypertension
VasodilationNeurotransmiters as histamine,bradykinin, or hormones like arginine-vasopressine (AVP) and norepinephrine (ΝΕ) acting in certain endothelial receptors or mechanic powers (shear stress) control ΝΟ production and circulation (EDRF)as long as other vasodilator substances like prostacyclin and endothelial dependent hyperpolarizator factor (EDΗF) histamine bradykinin endothelium bradykinin dilation Smooth muscles
Vasoconstriction Situations like growing old, BP, DM, atherosclerosis and menopause, can lead to the production and circulation of contractive factors from endothelial cells, like prostanoids, ΤΧΑ2, PGH2, free Ο2radicals, that neutralize the vasodilative NO and PGI2 action . activation endothelium activation Smooth muscles contraction
OTHER FACTORS CAUSING ARTERIAL HYPERTENSION Inherited factors Polymorphism 2 of ACE gene : salt sensitivity Polymorphism of a-adukine gene :increased Na-H countertransport. Smoking : through insulin resistance, increased endothelin and decreased dilation of vessel endothelium. Alcohol:through increased SNS activity, increased insulin resistance, thus hyperinsulinemia and alterations in cell membrane leading to increased intracellular calcium.