Download

1 / 8
80 likes | 217 Vues
This work presents a dynamic model for protein folding, utilizing a simplified representation that enhances computational efficiency. By applying a novel solvent model, our approach incorporates the effects of hydrophilic and hydrophobic sidechains while leveraging Langevin dynamics. The model addresses constraints imposed by secondary structure elements and hydrogen bonding, promoting accurate folding predictions. Features such as crowd control within a simulated cellular environment bolster the modeling of protein dynamics, providing valuable insights into protein stability and structure.

E N D
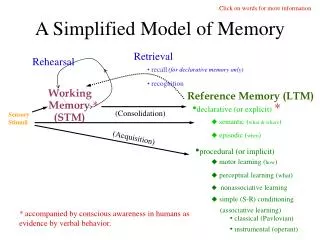
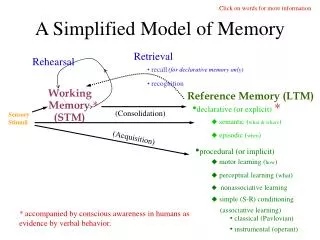
Poing A dynamic model of protein folding • Simplified representation • Fast • Novel solvent model • Extensible
Poing Small hydrophilic sidechain structure simplification Backbone C-alpha Protein backbone Large hydrophobic sidechain
Poing linear elastic springs
Poing Langevin dynamics is F = ma ... PLUS a model for solvent a: acceleration F: net force (sum of many forces) γ: drag factor v: current particle velocity R: randomly distributed vector m: particle mass
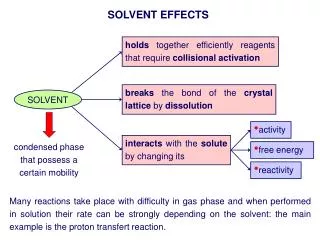
Poing Secondary structure and hydrogen bonding Solvent model leads tohydrophobic collapse Sidechain size and shape
Phyre + Poing HMM ARNDLSLDLVCS……. PSI-Blast Hidden Markov Model DB of KNOWN STRUCTURES HMM-HMM matching FINAL MODEL POING: Synthesise from virtual ribosome. Springs for constraints. Ab initio modelling of missing regions. Extract pairwise distance constraints
Protein Folding Requires Crowd Control in a Simulated Cell. Jefferys BR, Kelley LA and Sternberg MJE Journal of Molecular Biology (2010) Volume 397, Issue 5, 16 April 2010, Pages 1329-1338