Norm Conserving Pseudopotentials and The Hartree Fock Method
350 likes | 844 Vues
Norm Conserving Pseudopotentials and The Hartree Fock Method. Eric Neuscamman Mechanical and Aerospace Engineering 715 May 7, 2007. Goals. Produce a stand alone Hartree Fock code using C++ Apply code to generate pseudopotentials. Goals. Produce a stand alone Hartree Fock code using C++

Norm Conserving Pseudopotentials and The Hartree Fock Method
E N D
Presentation Transcript
Norm Conserving Pseudopotentialsand The Hartree Fock Method Eric Neuscamman Mechanical and Aerospace Engineering 715 May 7, 2007
Goals • Produce a stand alone Hartree Fock code using C++ • Apply code to generate pseudopotentials
Goals • Produce a stand alone Hartree Fock code using C++ • Apply code to generate pseudopotentials Success!
Goals • Produce a stand alone Hartree Fock code using C++ • Apply code to generate pseudopotentials Success! • Very limited success.
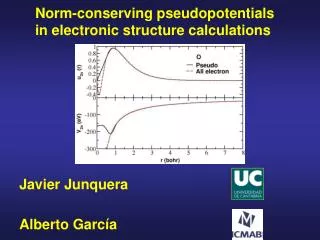
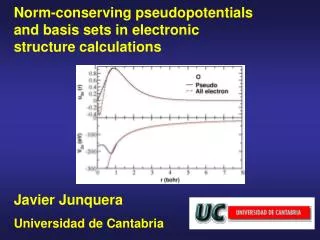
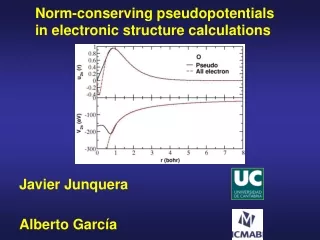
Basic Idea Pseudopotentials simplify calculations 1. By removing core electrons Carbon n = 6 n3 = 216 nv = 4 nv3 = 64 Silicon n = 14 n3 = 2744 2. By creating nodeless valence orbitals
Creating Pseudopotentials Step 1: Solve atomic system exactly. Step 2: Construct the target valence orbital. It should be exact for large r. Step 3: Invert the Schrodinger eq. for the V(r) that produces your valence orbital
The Hartree Fock Method Apply the variational principal to a Slater determinant. Result This differential equation has an infinite number of solutions.
Solving the Fock Equation Introduce a basis Restricted Hartree Fock: Assumes all orbitals are doubly occupied Unrestricted Hartree Fock:
Solving the Fock Equation The basis gives a matrix eigenvalue problem Diagonal Eigenvalue Matrix Fock Matrix Orbital Coefficients Overlap Matrix
Solving the Fock Equation Structure of the Fock operator One Electron Integrals Two Electron Integrals
Solving the Fock Equation Finding the integrals is the hardest part! One Electron Integrals Two Electron Integrals
Basis Sets To simplify integration, choose linear combinations of gaussian basis functions. This gives us We have reduced a 6-dimensional integral to a 2-dimensional integral An analytic integral
Guess the initial density matrices Calculate the Fock Matrix Diagonalize the two systems to find the coefficient matrices Update the density matrices The SCF Algorithm Solution Found No Converged? Yes
Target Valence Orbitals Now that we know the exact valence orbital functions, we may construct new valence orbitals that lack nodes. • Questions • How do we ensure they match in the valence region? • How do we enforce normalization? (Norm conservation) Answer: Method of Hamman, Schluter, and Chiang
is a cutoff function such that Example: Outside the core, . Thus solutions to the exact and modified Schrodinger equations with the same eigenvalues will be identical in the valence region. Introduce a cutoff function Holding the core electrons fixed, re-solve the Schrodinger (Fock) equation, but with a modified potential:
Introduce a cutoff function To generate our target valence orbital, we repeatedly solve the modified Fock equation, adjusting c until the eigenvalue of the nodeless solution is the same as for the exact orbital.
A Potential Problem Incorporating in the Hartree Fock method means modifying the one and two electron integrals. One Electron: Not a problem Two Electron: Breaks symmetry!
Solution: Only modify the OEI Rather than modifying the nuclear, coulomb, and exchange potentials, only modify the nuclear attraction. Then only the one electron integrals need modification and the method is tractable again.
Norm Conservation Due to the homogeneity of the Schrodinger equation, the modified and exact valence functions may differ by a constant multiple. This is easily fixed by scaling the modified function to match the exact function in the valence region. Afterwards, however, our function is no longer normalized!
Norm Conservation To re-normalize our function without altering its valence behavior, we must change it’s form in the core. This is achieved by using a cutoff function again. The normalization condition is then Of the two roots for δ, choosing the smaller one will produce a smoother wavefunction.
The Pseudopotential We have now generated a target valence orbital that is normalized and matches the exact orbital outside the core. Our pseudopotential is then whatever potential generates our target orbital. To find it, we invert the Fock equation. Solve for me!
Easier Said Than Done The integral form of the coulomb and exchange operators, coupled with the fact that not all the core electrons will be in spherically symmetric orbitals, make inverting this equation cumbersome. Currently, my code can only calculate Vpseudo when all of the occupied orbitals are spherically symmetric. This limits me to Lithium and Berillium. Yuck!
Results Hartree Fock code correctly predicts orbital occupations for 2nd row elements (need to check 3rd row). Small modification would allow d-orbital basis functions to be employed, permitting modeling of transition metals. However, accuracy will degrade as relativistic effects grow. Pseudopotential method successful for both Li and Be. Results reported here employ the 6-31G basis set. Substantial work needed to allow calculation of PPs for atoms with p electrons.
Conclusion Although implementing it was an excellent educational tool, the Hartree Fock is ill-suited for pseudopotentials Even if my code’s current shortcomings could be removed, relavistic effects would prevent the method from applying to heavier atoms where pseudopotentials can greatly reduce the number of electrons.