Download

1 / 33
340 likes | 740 Vues
Biological Motivation Gene Finding in Eukaryotic Genomes. Rhys Price Jones Anne R. Haake. Recall from our previous discussion of gene finding in prokaryotes:. The major strategies in gene finding programs are to look for: Signals/Features Content/Composition

E N D
Biological MotivationGene Finding in Eukaryotic Genomes Rhys Price Jones Anne R. Haake
Recall from our previous discussion of gene finding in prokaryotes: The major strategies in gene finding programs are to look for: • Signals/Features • Content/Composition • Similarity to known genes (BLAST!)
3 Major Categories of Information used in Gene Finding Programs • Signals/features = a sequence pattern with functional significance e.g. splice donor & acceptor sites, start and stop codons, promoter features such as TATA boxes, TF binding sites • Content/composition -statistical properties of coding vs. non-coding regions. • e.g. codon-bias; length of ORFs in prokaryotes; CpG islands GC content • Similarity-compare DNA sequence to known sequences in database • Not only known proteins but also ESTs, cDNAs
In Prokaryotic Genomes • We usually start by looking for an ORF • A start codon, followed by (usually) at least 60 amino acid codons before a stop codon occurs • Or by searching for similarity to a known ORF • Look for basal signals • Transcription (the promoter consensus and the termination consensus) • Translation (ribosome binding site: the Shine-Dalgarno sequence) • Look for differences in sequence content between coding and non-coding DNA • GC content and codon bias
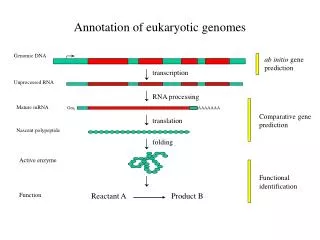
The Complicating factors in Eukaryotes • Interrupted genes (split genes) • introns and exons • Large genomes • Most DNA is non-coding • introns, regulatory regions, “junk” DNA (unknown function) • About 3% coding • Complex regulation of gene expression • Regulatory sequences may be far away from start codon
Some numbers to consider: • Vertebrate genes average about 30Kb long • varies a lot • Coding region is only about 1-2 Kb • Exon sizes and numbers vary a lot • Average is 6 exons, each about 150 bp long • An average 5’ UTR is about 750 bp • An average 3’UTR is about 450 bp • (both can be much longer) • There are huge deviations from all of these numbers • e.g. dystrophin is 2.4 Mb long ; factor VIII gene has 26 exons, introns are up to 32 Kb (one intron produces 2 transcripts unrelated to the gene!) • There are genes without introns: called single-exon or intronless genes
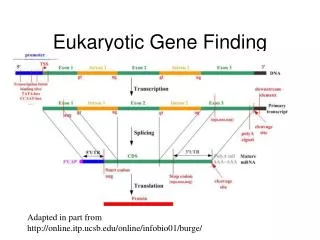
Eukaryotic Gene Structure www.bio.purdue.edu/courses/biol516/eukgenestructure.gif
Given a long eukaryotic DNA sequence: • How would you determine if it had a gene? • How would you determine which substrings of the sequence contained protein-coding regions?
In prokaryotic genomes we usually start by looking for ORFs. • Is this a good approach for the eukaryotic genome?
So, what’s the problem with looking for ORFs? “split” genes make it difficult to define ORFs • Where are the stops and stops? • What problems do introns introduce? • What would you predict for the size of ORFs? • (you can’t with any certainty!)
Most Programs Concentrate on Finding Exons • Exon: the region of DNA within a gene that codes for a polypeptide chain or domain • Intron: non-coding sequences found in the structural genes
Splice Sites used to Define Exons • Splice donor (exon-intron boundary) and splice acceptor (intron-exon boundary) • are consensus sequences • A statistical determination of the pattern;approximates the pattern • C(orA)AG/GTA(orG)AGT "donor" splice site • T(orC)nNC(orT)AG/G "acceptor" splice site • Example: http://www.library.csi.cuny.edu/~davis/Bioinfo_326/lectures/lect9_10/SpliceSite.htm
Gene finding programs look for different types of exon • single exon genes: begin with start codon & end with stop codon • initial exons: begin with start codon & end with donor site • internal exons: begin with acceptor & end with donor • terminal exons: begin with acceptor & end with stop codon
How are correct splice sites identified? • There are many occurrences of GT or AG within introns that are not splice sites • Statistical profiles of splice sites are used http://www.lclark.edu/~lycan/Bio490/pptpresentations/mutation/sld016.htm
Other Biologically Important Signals Used in Gene Finding Programs • Transcriptional Signals • Transcription Start: characterized by cap signal • A single purine (A/G) • TATA box (promoter) at –25 relative to start • Polyadenylation signal: AATAAA (3’ end) • Major Caveat: not all genes have these signals • Makes it difficult to define the beginning and end of a gene
Upstream Promoter Sites • Transcription Factor (TF) sites • Transcription factors are sequence-specific DNA-binding proteins • Bind to consensus DNA sequences • e.g. CAAT transcription factor and CAAT box • Many of these • Vary in sequence, location, interaction with other sites • Further complicates the problem of delineating a “gene”
Translation Signals • Kozak sequence • The signal for initiation of translation in vertebrates • Consensus is GCCACCatgG • And of course.. • Translation stop codons
Codon Biasin Eukaryotic Genomes • Yeast Genome: arg specified by AGA 48% of time (other five equivalent codons ~10% each) • Fruitfly Genome: arg specified by CGC 33% of time (other five ~13% each) • Complete set of codon usage biases can be found at: • http://www.kazusa.or.jp/codon/CUTG.html
GC Content in Eukaryotes • Overall GC content does not vary between species as it does in prokaryotes • GC content is still important in gene finding algorithms • CpG Islands • CG dinucleotides occur at low frequency overall in the genome • Exception: CpG islands near promoters • CG dinucleotides occur at level predicted by chance • -1,500 to +500 (relative to transcription start site)
CpG Islands • Occurrence related to methylation • Methylation of C in CG dinucleotides • Methylation of C makes CpG prone to mutation (e.g. to TpG or CpA) • Level of methylation is low in actively transcribed genes • Transcription requires a methyl-free promoter
Gene Finding Strategies • Homology-based approach • Find sequences that are similar to known gene sequences • ab initio-based approach is to identify genes by: • Signal sequences • Composition
Gene Finding Input DNA sequence (string of G’s, A’s, C’s, T’s) Known gene(s)? BLAST Known gene No High score, above the threshold? CpG Islands? Low score Low score High score, above the threshold? Promoter ? High score, above the threshold? Low score ORF Signals ? Splice Sites ? Low score High score, above the threshold? Probability of being a gene. Low cumulative score High cumulative score
List of Gene Finding Programs • http://www.hgc.ims.u-tokyo.ac.jp/~katsu/genefinding/programs.html • http://www.hku.hk/bruhk/sggene.html
Homology-Based Approaches in Eukaryotic Genomes • More complicated than prokaryotes due to split genes • Genome sequence -> first identify all candidate exons • Use a spliced alignment algorithm to explore all possible exon assemblies & compare to known • e.g. Procrustes • Limitations: • must have similar sequence in the database with known exon structure • Sensitive to frame shift errors
Procrustes • Gene Recognition via spliced alignment • Given a genomic sequence and a set of candidate exons, the spliced alignment algorithm explores all possible exon assemblies and finds a chain of exons with the best fit to a related target protein • http://hto-13.usc.edu/software/procrustes/#salign
GenScan • Allows integration of multiple types of information • Earlier programs considered features of gene structure in isolation • Uses a generalized HMM (one state might use a weight matrix model, another an HMM) http://genes.mit.edu/GENSCAN.html
GenScan Probabilistic Model of Genes • Accounts for many of the known structural & compositional properties of genes including: • typical gene density • typical number of exons per gene • distribution of exon sizes for different types of exon • compositional properties of coding vs. non-coding • translation initiation (Kozak) • termination signals • TATA box, cap site and poly-adenylation signals • donor and acceptor splice sites
GenScan • Uses as a training set 238 multi-exon genes and 142 single-exon genes from GenBank to compute parameters • Initial state probabilities • Transition probabilities • State length distributions • Probabilistic models for the states • The states correspond to different functional units on a gene e.g promoter regions, exon • Transitions ensure that the order that the model marches through the states is biologically consistent • Length distributions take into account that different functional units have different lengths.
GenScan • Signal models used by GenScan - WMM= weight matrix model for transcriptional and translational signals (translation initiation, polyadenylation signals, TATA box etc.) e.g. polyadenylation signal is modeled as a 6 bp WMM with AATAAA as the consensus sequence (uses annotated data from GenBank) -WAM= weight array model; assumes some dependencies between adjacent positions in the sequence e.g. used for the pyrimidine-rich region and the splice acceptor site -Maximal dependency decomposition e.g. used for donor splice sites
GenScan • does not use similarity search • uses double stranded genomic sequence model • potential genes on both strands are analysed simultaneously • Limitations: • cannot handle overlapping transcription unit • does not address alternative splicing
GRAIL • GRAIL (Gene Recognition and Assembly Internet Link) • uses a number of sensor algorithms to evaluate coding potential of a DNA sequence • features include 6-mer composition, GC composition and splice junction recognition • the output of the sensor algorithms is input to a neural network, which uses empirical data for training.
GRAIL-exp • http://compbio.ornl.gov/grailexp/gxpfaq1.html