Cystic Fibrosis
470 likes | 946 Vues
Cystic Fibrosis. S4S. Definition. Common autosomal recessive disorder, characterised by: Chronic broncho-pulmonary infection Pancreatic insufficiency Subsequent multi-system complications. Aetiology & Epidemiology. Most common inherited disease in white races

Cystic Fibrosis
E N D
Presentation Transcript
Cystic Fibrosis • S4S
Definition • Common autosomal recessive disorder, characterised by: • Chronic broncho-pulmonary infection • Pancreatic insufficiency • Subsequent multi-system complications
Aetiology & Epidemiology • Most common inherited disease in white races • 1 in 25 white Europeans are carriers • 1 in 2,500 children are affected • Defective/missing CFTR from q arm of chromosome 7 • DF508 mutation accounts for 75% of CF genes in UK
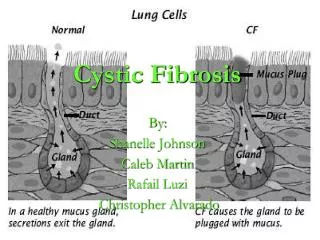
Pathophysiology • Faulty CFTR does not allow ions to leave cells • Reduced lumen [chloride] • Increased cellular sodium reabsorption from lumen • Reduced excretion of water • Hyper-viscous mucus (blocks pancreatic ducts) • Bacterial DNA exacerbates this problem (hence DNAses) • NB. Normal CTFR allows ion reabsorption from sweat
CFTR • Left: Unaffected channel • Ions flow freely • Right: CTFR in CF • Channel is blocked • Mucus accumulates
Signs and Symptoms • Later diagnosis = greater array of S&S • Typically detected at 0-2 years due to: • Recurrent infection • Large foul smelling stools • Failure to thrive
Presenting at Birth: • Failure to thrive • Recurrent chest infections • Bronchiectasis • Meconium ileus 10-20%
<2 years • Also: • Hx of prolonged neonatal jaundice • Steatorrhoea
2-8 years • Also: • Bronchiectasis • Rectal prolapse • Nasal polyps • Sinusitis
8 years+ • Also: • Cor pulmonale (^lung pres.) • T2DM • Cirrhosis • Intestinal obstruction • Pneumothorax • Haemoptysis • Infertility • Osteoporosis • Psychological disorders
Common signs • Chest hyperinflation • Nasal polyps • Cyanosis • Hepatomegaly • Expiration wheeze • Inspiratory crepitations • Air trapping • Chronic inflammation • Reduced oxygen transfer • Blocked hepatic ducts • Inflammation • Reduced compliance
Investigations • Sweat test • Chloride & Sodium measured (Cl>Na) • Children normally <60mmol/L • Adults normally <90mmol/L • CXR - upper lobe bronchiectasis, later diffuse • Faecal Elastase • Diagnostic of pancreatic insufficiency (<200mcg/g of stool) (3%/3-8%) • Guthrie • IRT raised in babies with CF; sweat test to confirm and screening for genetic mutations
Complications • Many and varied • Respiratory • Digestive • Reproductive • Cardiac • Hepatic • Biliary
Complications • Why would you get: • Nasal Polyps • Meconium ileus • Vitamin deficiency • Cor pulmonale • Haemoptysis/pneumothorax • Inflammation/oedema • Pancreatic insufficiency • Reduced absorption • Raised pulm. pressure • Infection, inflammation and coughing trauma
Complications: Important (PLOD) • Respiratory: • Initially Staph and HIB infections, later susceptible to Pseudomonas Aeroginosa • Severe asthma and unexplained Bronchiectasis • CF related Liver disease • Osteoporosis due to malnutrition and steroids • Diabetes due to scarring and steroids
Alternate Diagnosis • Failure to thrive • Celiac • Pancreatic insufficiency disorder from other causes (
Management: Considerations • Cystic Fibrosis centres ideal with MDT • Baseline assessment and investigations (spirometry) • Regular outpatient follow-up • Parent and patient education • No cure! • Goals of treatment: • Maintain adequate lung function • Maintain adequate growth • Manage complications
Respiratory Management • Prophylactic flucloxicillin (6 week cycling reduces resistance) • IV Abx for pseudomonas (can be at home) • Will likely become resistant eventually • Avoid contact with other pseudomonas patients • Regular physiotherapy • Nebulised saline/antibiotics • CPAP & home oxygen • Lung Transplant
Nutrition Management • Strong correlation between well-managed nutrition and wellbeing in CF • Caloric excess (NG in extremes) • Pancreatic enzymes • PPIs to aid enzyme function • DO NOT restrict fat intake • ADEK supplementation • Aim for normal growth
Hepatic Management • UDCA (ursodeoxcholic acid) to prevent impairment • Liver transplant in severe cirrhosis
Diabetes & Osteoporosis • Insulin for Diabetes • Vitamin D supplementation • Calcium Supplementation if low BMD • Encourage weight bearing activities
Prognosis • Dependent on severity • Most live to 30s now • Estimated that 80% born today will live to mid 40s • Double lung transplant can increase life expectancy by 10 years