Download

1 / 15
150 likes | 776 Vues
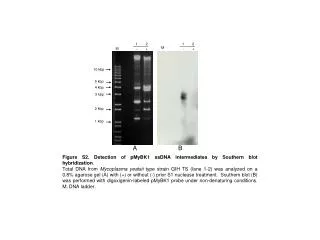
SOUTHERN BLOT Capillary Transfer of DNA to a Membrane. ABE WORKSHOP JUNE 6-24, 2005. Purpose. Detection of specific sequences among DNA fragments separated by gel electrophoresis. Restriction fragments appear as a smear rather than as discrete bands on the gel.

E N D
SOUTHERN BLOTCapillary Transfer of DNA to a Membrane ABE WORKSHOP JUNE 6-24, 2005
Purpose • Detection of specific sequences among DNA fragments separated by gel electrophoresis. • Restriction fragments appear as a smear rather than as discrete bands on the gel. • When the DNA is transferred to the membrane the fragments retain the same pattern of separation they had on the gel but appear as bands.
Method • Submerge the electrophorized gel in 0.2N HCl and agitate gently on a platform shaker until the blue band turns green. • Pour off the HCl and then rinse the gel several times with deionized water. • Cut off the top right corner of the agarose gel to simplify orientation during succeeding operations. • Soak the gel in Denaturation Buffer for 30 minutes with gentle agitation. • Rinse the gel with dH2O then soak in Neutralization Buffer for 30 minutes. • Cut 3 pieces of (Whatman) filter paper to the size of the gel and 1 piece long enough to reach the bottom of the inverted gel tray.
Cut 1 piece of positively charged nylon membrane to the exact size of the gel. Cut off the top right corner of the membrane to match the gel. • While the DNA is denaturing, construct the following system: • Invert gel tray in a large glass baking dish with the long piece of filter paper over it. The ends of the filter paper should drop to the bottom of the dish. • Fill the dish with Transfer Buffer (20X SSC). • Place 1 small piece of filter paper (wet with Transfer Buffer) on top of the gel tray, smooth out all air bubbles with a glass rod. • Remove the gel from the Neutralization Buffer and place it inverted on the filter paper. Make sure there are no air bubbles between the blotting paper and the gel.
Surround but do not cover the gel, with saran wrap or Parafilm. • Wet the top of the gel with more Transfer Buffer. • Soak the nylon membrane in dH2O and place on top of the gel so that the cut corners are aligned. To avoid bubbles, touch one corner of the membrane to the gel and gently lower the membrane onto the gel. • Place the two filter papers on top of the membrane. Smooth away any air bubbles. • Cut or fold a stack of paper towels (5-8 cm. high) and place them on top of the two filter papers. • Place a glass plate and a beaker containing water on top of the stack to weigh the stack down. • Allow the transfer of DNA to continue for 8-24 hours.
The next day, remove the weight, glass plate and paper towels. • Lift the filter paper, the nylon membrane and the gel together, then invert. • Mark the positions of the gel wells on the membrane with a soft lead pencil. • Peel the membrane from the gel and rinse in 6X SSC for 10 minutes • Wrap membrane in saran wrap and place it in the UV crosslinker for 2 minutes to crosslink DNA to membrane. • Roll nylon membrane into a small roll and place inside a roller tube with the side containing the DNA facing the inside.
Preparing probe: Combine 10 µl template DNA & 6 µl dH2O. Denature the DNA by heating in a boiling water bath for 10 minutes and quickly chilling on ice. Mix DIG-High Prime thoroughly and add 4 µl to the denatured DNA, mix & centrifuge briefly. Incubate for 1 hour at 37ºC. Stop the reaction by adding 2 µl 0.5M EDTA. • Add 15 ml DIG Easy Hybrid probe to the roller tube. • Place the tube in the hybridizer for 30 minutes at 45ºC.
Discard probe. Add a preheated mixture of 4 ml DIG Easy Hybrid and 10 ml DNA to the tube. • Put the tube back into the hybridization chamber at 65ºC overnight. • The next day pour off the probe. • Wash membrane twice for 5 min. each time in 2X Wash solution at room temperature with gentle rotation. • Wash membrane twice for 5 min. each time in prewarmed 0.1X or 0.5X Wash solution at 68ºC. • Equilibrate membrane in Wash buffer for 2 min. at room temperature. • Incubate membrane in 30 ml Maleic buffer for 30 min. at room temperature with gentle rotation.
Remove membrane, add 3 µl Anti-Digoxigenin-AP, Fab Fragments, replace membrane and rotate gently for 30 minutes. • Wash membrane twice for 15 min. each time in Wash buffer. • Equilibrate membrane in Detection buffer for 2 min. • Place membrane (nucleic acid side up) on saran wrap. Spot 1 ml CSPD/Detection buffer solution over the membrane. Cover with another layer of saran wrap and seal the edges. Be sure no air bubbles are present between membrane and saran wrap. Incubate for 5 min. • Tape membrane into pre-warmed x-ray cassette and incubate at 37ºC for 10 minutes.
In the darkroom under a safelight, cut autorad x-ray film to appropriate size and tape over membrane Close cassette and expose for 15 minutes. • Remove the film from the cassette under a safelight. Develop for 1 minute, fix for 3 minutes, then rinse twice with water and hang to dry.
Results • No banding is seen although the gel showed presence of DNA. • Probe may not have been good. • Exposure times may have been incorrect (only 5 min. & overnight were used due to time constraints).
PCR & GEL ELECTROPHORESIS • Prepare 1 PCR tube for each sample as follows • 32.3 ul d. water • 5 ul template DNA (1ng/mL) • 5 ul 19X PCR Buffer • 3 ul 25 mM MgCl • 2.5 ul 4 mM dNTP • 1.0 ul left primer (20 uM) • 1.0 ul right primer (20 mM) • 0.2 ul Taq DNA polymerase
Method • Load PCR machine and run a standard cycle. • 95ºC for 5 minutes to completely denature DNA • Then 35 cycles as follows: • 1 minute at 95ºC (denaturing) • 1 minute at 50ºC (annealing) • 1 Minute at 72ºC (extension) • After PCR is complete remove samples and run them on a gel.
ResultsPCR products & Vanilla DNA • Lane 1: 1 kb DNA ladder • Lane 2: Vanilla DNA • Lane 3: Blank • Lane 4: Pds Red template • Lane 5: Aco template • Lane 6: PSBA template