Download
1 / 25
250 likes | 268 Vues
Modeling the Structures of Proteins and Macromolecular Assemblies. Depts. Of Biopharmaceutical Sciences and Pharmaceutical Chemistry California Institute for Quantitative Biomedical Research University of California at San Francisco. Andrej Š ali http://salilab.org/. 3/25/03.

E N D
Modeling the Structures of Proteins andMacromolecular Assemblies Depts. Of Biopharmaceutical Sciences and Pharmaceutical Chemistry California Institute for Quantitative Biomedical Research University of California at San Francisco Andrej Šali http://salilab.org/ 3/25/03
Yeast and E. coli ribosomes: Electron microscopy, comparative modeling, and structural genomics. • Yeast Nuclear Pore Complex: Low-resolution modeling of large assemblies; bridging the gaps between structural biology, proteomics, and system biology. • Comments. 4/6/03
S. cerevisiae ribosome Fitting of comparative models into 15Å cryo- electron density map. 43 proteins could be modeled on 20-56% seq.id. to a known structure. The modeled fraction of the proteins ranges from 34-99%. C. Spahn, R. Beckmann, N. Eswar, P. Penczek, A. Sali, G. Blobel, J. Frank. Cell107, 361-372, 2001. 3/25/03
E. coli ribosome H.Gao, J.Sengupta, M.Valle, A. Korostelev, N.Eswar, S.Stagg, P.Van Roey, R.Agrawal, S.Harvey, A.Sali, M.Chapman, and J.Frank. Cell, in press. UponEF-G binding, the ribosome becomes less compact. In contrast to mRNA, many protein contacts undergo large conformational changes, suggesting ribosomal proteins facilitate the dynamics of translation. 4/6/03
Select templates using permissive E-value cutoff 1 MODPIPE:Large-Scale Comparative Protein Structure Modeling START 1 Get profile for sequence (NR) Expand match to cover complete domains PSI-BLAST Scan sequence profile against representative PDB chains Align matched parts of sequence and structure MODELLER For each template structure For each target sequence Scan PDB chain profiles against sequence Build model for target segment by satisfaction of spatial restraints Evaluate model END R. Sánchez & A. Šali, Proc. Natl. Acad. Sci. USA 95, 13597, 1998. N. Eswar, M. Marti-Renom, M.S. Madhusudhan, B. John, A. Fiser, R. Sánchez, F. Melo, N. Mirkovic, A. Šali. 3/25/03
http://salilab.org/modbase Pieper et al., Nucl. Acids Res. 2002. 3/25/03
70% of models based on <30% sequence identity to template. On average, only a domain per protein is modeled (an “average” protein has 2.5 domains of 175 aa). Comparative modeling of the TrEMBL database Unique sequences processed: 733,239 Sequences with fold assignments or models: 415,937(57%) 4/03/02 ~4 weeks on 500 Pentium III CPUs 3/25/03
Ca equiv 90/134 RMSD 1.17Å Ca equiv 147/148 RMSD 0.41Å Ca equiv 122/137 RMSD 1.34Å Sidechains Core backbone Loops Sidechains Core backbone Loops Alignment Fold assignment Sidechains Core backbone Loops Alignment Model Accuracy Marti-Renom et al. Annu.Rev.Biophys.Biomol.Struct. 29, 291-325, 2000. HIGH ACCURACY LOW ACCURACY MEDIUM ACCURACY NM23 Seq id 77% CRABP Seq id 41% EDN Seq id 33% X-RAY / MODEL 4/6/03
Future directions Make sure we have building blocks (structural genomics). Develop methods for simultaneous fitting of proteins into EM density and conformational modeling (induced fit, comparative modeling, ab initio). Need large computing (eg, cluster of hundreds of nodes with >1GB memories). 4/6/03
Modeling of the yeast nuclear pore complex by satisfaction of spatial restraints (MODELLER) F. Alber*, T. Suprapto, J. Kipper, W. Zhang, L. Veenhoff, S. Dokudovskaya, M. Rout, B. Chait, A. Šali* Rockefeller University, New York *UCSF 4/6/03
Modeling macromolecular assemblies by satisfaction of spatial restraints • Representation of a system. • Scoring function (spatial restraints). • Optimization. There is nothing but points and restraints on them. Sali, Ernst, Glaeser, Baumeister. From words to literature in structural proteomics.Nature 422, 216-225, 2003. 3/25/03
Modeling of NPC • Stochiometry. • Parse proteins into domains. • Protein and “sub-complex’ shapes from Stokes radii. • Excluded volume of proteins. • Symmetry of NPC (EM). • Radial and axial localization of proteins (IEM). • Protein-protein proximity (immuno-purification). • Binary protein-protein contacts from “overlay” experiments. • Modeling in the context of the nuclear envelope. • Comparative models for some domains. • Structural genomics of nucleoporins. 4/6/03
Schematic structure of yeast NPC C.W. Akey, M. Rout TOP VIEW SIDE VIEW cytosolic side nuclear membrane nuclear side half-spoke contains ~30 nucleoporin proteins (NUPs). ~480 NUPs in NPC. half-spoke spoke 3/25/03
complex dimension f(res) [A] [sum of residues] Complications: • multiple protein copies in half-spoke • inter-spoke interactions. Protein-protein “contacts” For each protein pair within a half-spoke, the upper bound on the center-center distance is the estimated maximal complex diameter: 3/25/03
3/25/03 11/18/02 Successful optimization
NUP85 SEH1 Scan of figure Cdc31 NUP120 NUP84 NUP145C Rappsilber J, Siniossoglou S, Hurt EC, Mann M. Anal Chem 2000 Jan 15;72(2):267-75. NUP84-complex Experiment Model 4/6/03
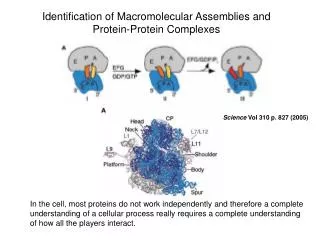
Structural proteomics aims to characterize structures of most macromolecular complexes, in space and time. On average, a domain may interact with a few other domains. The function of a complex is determined by its structure and dynamics. There are too many complexes to be determined directly by high-resolution experimental structure determination. Thus, just like in structural genomics, an efficient combination of experiment and computation is required. 4/6/03
Structural Proteomics versus Structural Genomics Potential targets not clear clear Target selection not clear clear Scope not clear clear Structure determination hybrid X-ray or NMR Functional Annotation essential not a major focus There are additional sciences and technologies in structural proteomics, relative to structural genomics. 4/6/03
Characterize most proteinsequencesbased on related knownstructures: The number of “families” is much smaller than the number of proteins. Any one of the members of a family is fine. Sali. Nat. Struct. Biol. 5, 1029, 1998. Sali et al. Nat. Struct. Biol., 7, 986, 2000. Sali. Nat. Struct. Biol.7, 484, 2001. Baker & Sali. Science 294, 93, 2001. Structural Genomics Characterize most protein sequences based on related known structures. There are~16,00030% seq id families (90%) (Vitkup et al. Nat. Struct. Biol. 8, 559, 2001) 3/25/03
Target selection for structural proteomics(scope) Comprehensive coverage. What targets: Stable assemblies? Transient complexes? Pairs of domains? Can they be organized into groups? How many such groups are there? 4/6/03
Target selection for structural proteomics? • There are ~3,000 folds containing ~90% of all sequences. • A target: Binary domain-domain interface. • There may be a finite number of domain-domain interfaces, largely defined by the domain fold types. • Given pairwise interactions, a large assembly may be reconstructed. 4/6/03
Protein and assembly structure by experiment and computation Sali, Ernst, Glaeser, Baumeister. From words to literature in structural proteomics. Nature 422, 216-225, 2003. 3/25/3 3/25/03
Modeling macromolecular assemblies by satisfaction of spatial restraints • Representation of a system. • Scoring function (spatial restraints). • Optimization. There is nothing but points and restraints on them. Sali, Ernst, Glaeser, Baumeister. From words to literature in structural proteomics.Nature 422, 216-225, 2003. 3/25/03
Future directions • Development of general/flexible/hierarchical representation of assemblies. • Quantifying spatial information from experiments. • Optimization of structure. • Toy models. • Space, time. 4/6/03
Role of NIH • Structural proteomics is timely and feasible. • Humongous integration of concepts, sciences, methods, tools, people. • Thus, need researchcenters, but also “R01” research. • Significant computing. • Bridging the gaps between structural biology, proteomics, and system biology. 4/6/03