Download
1 / 16
160 likes | 288 Vues
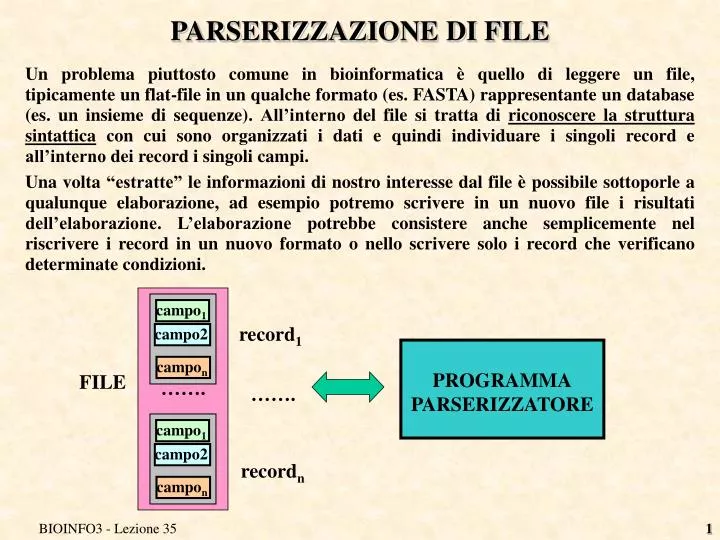
campo 1. campo 1. campo2. campo2. campo n. campo n. ……. PARSERIZZAZIONE DI FILE.

E N D
campo1 campo1 campo2 campo2 campon campon ……. PARSERIZZAZIONE DI FILE • Un problema piuttosto comune in bioinformatica è quello di leggere un file, tipicamente un flat-file in un qualche formato (es. FASTA) rappresentante un database (es. un insieme di sequenze). All’interno del file si tratta di riconoscere la struttura sintattica con cui sono organizzati i dati e quindi individuare i singoli record e all’interno dei record i singoli campi. • Una volta “estratte” le informazioni di nostro interesse dal file è possibile sottoporle a qualunque elaborazione, ad esempio potremo scrivere in un nuovo file i risultati dell’elaborazione. L’elaborazione potrebbe consistere anche semplicemente nel riscrivere i record in un nuovo formato o nello scrivere solo i record che verificano determinate condizioni. record1 PROGRAMMA PARSERIZZATORE FILE ……. recordn BIOINFO3 - Lezione 35
FILE FASTA • Supponiamo che il file da analizzare sia in formato FASTA, ad esempio un file di sequenze EST scaricato da GENBANK. Individuamo nei record i campi di nostro interesse 1 2 3 4 5 1) Il numero identificativo gi del record genbank 2) Il codice accession 3) Il nome della EST 4) La descrizione della EST 5) La sequenza della EST BIOINFO3 - Lezione 35
1 2 3 4 5 ELABORAZIONE • L’elaborazione che ci interessa effettuare su ogni singolo record è quella di inserire un record corrispondente all’interno di un database relazionale SQL di sequenze EST. Le 5 informazioni estratte da ogni record dovranno produrre un corrispondente comando di insert MySQL insert into est (gi,accession,nome,descrizione,sequenza) values(8777287,’AB044776’,’AB044776’,’Panax…’,‘GAAGAA…’) BIOINFO3 - Lezione 35
2 ALTERNATIVE • Abbiamo visto che esistono due modi alternativi per interagire con un database SQL e di conseguenza è possibile eseguire l’operazione di insert in due modi alternativi • Dalla linea di comando del client MySQL • Da programma (embedding) • Nel primo caso il programma di parserizzazione creerà un file di comandi SQL che poi daremo direttamente in pasto all’interprete MySQL. • Nel secondo caso le istruzioni di insert sono eseguite una ad una direttamente all’interno del programma, man mano che vengono letti i singoli record. • La seconda soluzione sembrerebbe più “automatizzata”, però anche nel primo caso, una volta scritto completamente il file, è possibile far eseguire da programma (con una istruzione qx o system) il comando UNIX di lettura ed esecuzione del file da parte del client mysql BIOINFO3 - Lezione 35
campo1 campo1 campo1 campo1 campo2 campo2 campo2 campo2 campon campon campon campon ……. ……. SCHEMATIZZANDO… • Proveremo a vedere entrambe le soluzioni in quanto la parte di programma che cambia è minima (evidenziata in rosso) while (riconoscimento di un record){ estrazione campi definizione comando di insert scrittura del comando sul file } esecuzione del file 1) file di comandi SQL file parserizzatore while (riconoscimento di un record){ estrazione campi definizione comando di insert esecuzione del comando } SERVER MYSQL 2) DB BIOINFO3 - Lezione 35
5 4 1 2 3 LA STRATEGIA PER PARSERIZZARE IL FILE Abbiamo detto che la parserizzazione deve essere guidata dalla struttura sintattica del file. In Perl una possibilità è quella di leggere l’intero file ed assegnarlo ad un’unica stringa (che può contenere file anche molto grandi) e quindi procedere all’individuazione dei record. I record sono chiaramente separabili grazie al pattern “>gi|” posto all’inizio di ogni record. Si potrà pertanto effettuare una operazione di split basata proprio su questo pattern, ottenendo un array contenente tutti i record. Per ogni record si potrà effettuare successivamente un pattern matching per estrarre i 5 campi. >gi| lettura file split array ( , , ) >gi| pattern matching >gi| file stringa BIOINFO3 - Lezione 35
IL PARSERIZZATORE BIOINFO3 - Lezione 35
ESECUZIONE Per evitare di “sorbirsi” tutti i cicli di lettura fissiamo un break point! BIOINFO3 - Lezione 35
ESECUZIONE Lo split crea 22 record, ma il primo è vuoto (perché il file iniziava con il pattern di split). Per questo ho aggiunto il test if ($record) BIOINFO3 - Lezione 35
ESECUZIONE Prima di eliminare tutti gli spazi e i new-line dalla sequenza proviamo a vedere tutte le stringhe catturate dal pattern matching BIOINFO3 - Lezione 35
ESECUZIONE Vediamo la sequenza $seq prima e dopo la sostituzione del pattern Si può anche notare che l’operazione di pattern substitution azzera le variabili speciali $1, $2,… In generale ogni operazione sui pattern cancella i risultati della precedente BIOINFO3 - Lezione 35
ESECUZIONE Si procede quindi con il riconoscimento del secondo record e così via. Alla fine di ogni ciclo abbiamo a disposizione nelle variabili $gi, $ac, $nome, $descr, $seq tutti i campi per quel record BIOINFO3 - Lezione 35
CREAZIONE DI FILE DI COMANDI SQL Non ve l’avevo ancora detto (anche perché a fine programma i file aperti vengono comunque chiusi automaticamente) ma è buona norma chiudere un file quando è stato letto o scritto completamente BIOINFO3 - Lezione 35
ESECUZIONE BIOINFO3 - Lezione 35
ESECUZIONE DEL FILE DI COMANDI • Una volta scritti tutti i comandi SQL e chiuso il file di output ($nomefile.mysql) si può eseguire l’istruzione Perl: • qx{mysql btbm-xx <$nomefile.mysql}; • Dove btbm-xx è il nome del database su cui far eseguire i comandi. Il comando UNIX eseguito da Perl fa leggere ed eseguire al client mysql il file di comandi appena creato. • Lo stesso comando UNIX potrà anche essere dato non da programma ma a mano al termine dell’esecuzione del programma parserizzatore BIOINFO3 - Lezione 35
COMANDI SQL DA PROGRAMMA BIOINFO3 - Lezione 35