Download

1 / 31
320 likes | 449 Vues
Cerebral Palsy (CP) is a non-progressive neurological disorder that affects movement and posture, with an incidence of 1.9 to 2.3 per 1,000 live births. It often results from prenatal brain abnormalities rather than perinatal asphyxia. Key clinical manifestations include delayed motor development, abnormal muscle tone, and persistent primitive reflexes. Diagnosis is primarily through neurologic examination and history. Management aims to enhance locomotion, communication, and social integration through physical therapy, education, and supportive devices. Early intervention is critical for improving outcomes.

E N D
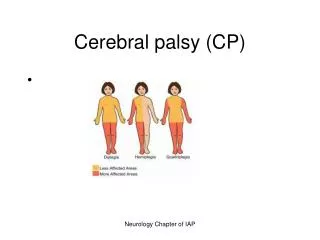
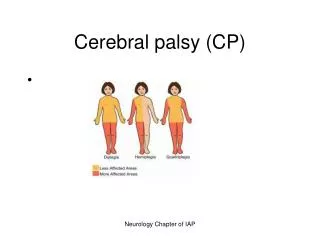
Cerebral Palsy (CP) Disorders characterized by early onset and impaired movement and posture. It is a non-progressive disease.
Incidence • 1.9-2.3 in every 1000 live births • in prevalence since the 1960’s due largely to the improved survival of VLBW infants.
Etiology • Greatest prevalence is seen in prematurely delivered infants. • Formerly thought to be R/T perinatal birth asphyxia, but now it is known that CP more commonly results from existing prenatal brain abnormalities. • 24% of cases have no identifiable cause.
Pathophysiology • Difficult to determine exactly • ANOXIA is most significant factor to cause pathologic brain damage. This is often 2ndary to other etiology. • The area of the lesion in the brain mostly determines the subsequent pathology.
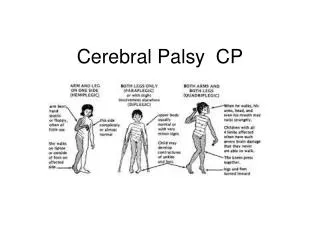
Clinical Classification of CP(see box 40-2 in Wong, p.1693 9th ed.)
Clinical Manifestations ( Box 40-4, Wong, p. 1695 9th ed.—Possible Signs) • Delayed gross motor development—universal manifestation of CP • Especially significant if other developmental behaviors e.g. speech & personal social are normal • Abnormal motor performance—Early sign is preferential unilateral hand use that may be apparent at ~6months of age.May stand or walk on toes
Clinical Manifestations (Box 40-4, Wong, p. 1695 9th ed. —Possible signs) • Alterations of Muscle Tone— • or resistance to passive movements is a sign of abnormal muscle tone. • Child may exhibit opisthotonic postures and stiffness on handling, dressing, or diapering. • Abnormal Postures— • From an early age, a child lying in a prone position will maintain the hips higher than the trunk with the legs and arms flexed or drawn under the body. • Spasticity may be mild or severe.
Clinical Manifestations (Box 40-4, Wong, p. 1695 9th ed. —Possible signs) • Reflex abnormalities— • Persistence of primitive infantile reflexes is one of the earliest clues to CP. • Associated Disabilities and problems— • Intellectual impairment—possible, but 70% are WNL • ADHD—poor attention span, marked distractibility, hyperactive behavior,and defects of integration • Seizures—most common in postnatal acquired hemiplegia • Drooling, feeding and speech needs, risk of aspiration & possible inadequate gas exchange. • Orthopedic complications • Constipation • Dental caries, malocclusion, gingivitis • Nystagmus, amblyopia & hearing loss
Diagnostic Evaluation • Neurologic Examination & History are the primary modalities for diagnosis • Recognizing etiologic factors that put the infant at risk is critical in the assessment and diagnostic process.
Therapeutic Management: General Concepts Box 40-5 p. 1696 9th ed.Therapeutic Interventions for CP • Broad aims: • Establish locomotion, communication, and self-help • Gain optimum appearance & integration of motor functions • Correct associated defects as effectively as possible • Provide educational opportunities adapted to the needs and capabilities of the individual child • Promote socialization experiences with other affected and unaffected children.
What is done— • Mobilizing devices—braces, crutches, wheelchairs, walkers • Surgery—when spasticity causes further deformities • Medication—drugs to spasticity are often NOT helpful in CP. Antianxiety meds may help child with athetosis.Skeletal muscle relaxants e.g. baclofen, methocarbamol (Robaxim), or dantrolene (Dantrium) & Valium may help short-term for older children & adolescents. Antiepileptic meds, e.g. phenobarbital & phenytoin are used routinely for children with seizures & CP.
What is done—(cont-d) • Technical aids—e.g. electromechanical toys that use biofeedback; microcomputers combined with voice synthesizers, or activated with a head-stick, tongue,or other voluntary muscle movement over which the child has control. • Other Considerations—care of vision & hearing deficits as well as dental care is essential early on.
Therapeutic Management: Therapies, Education, Recreation • Physical Therapy—one of the most commonly used conservative txmodalites. Involves PT, family, and nsg • Functional & Adaptive Training (Occupational Therapy)—training in manual skills and ADL’s must be started early • Speech Therapy—start early to prevent speech problems. • Education—individualize to the needs of the child • Recreation—sports, physical fitness, & recreation programs are encouraged for children with CP
Nursing Care (see Care Plan pp.1702-1703 in 9th ed.) • Reinforce therapeutic plan/assist in Normalization • Address Health Maintenance needs • Watch for fatigue, nutritional needs, safety needs • Support family • Help them cope with the emotional aspects of the disorder • Make appropriate referrals to support groups e.g United Cerebral Palsy Association. http://www.ucpa.org/search.cfm • Support hospitalized child— • the nurse’s actions should convey acceptance, affection, and friendliness and promote a feeling of trust and dependability.
Muscular Dystrophy Gradual degeneration occurs in muscle fibers progressive weakness and symmetric wasting away of skeletal muscle
3 Types of DystrophyTable 40-2; p. 1725 9th ed. • Pseudohypertophic (Duchenne) • X-linked Recessive • 1-3 years of age • Lordosis, Waddling gait • Rapid progression— • Death 15-25 after onset • Website Part 1 with newest Guidelines from MDA—12/09 • Website Part 2 with newest multidisciplinary guidelines from MDA—12-09
Limb-Girdle • Onset after 8 y/o • Weakness of proximal muscles of pelvic and shoulder girdle • Slow progression • Incapacitated 20 years after onset • OR slight disability
Facioscapulohumeral(Lansouzy-Dejerine) • Early adolescence • Symptoms • Lack of facial mobility • Can’t raise arms over head • Shoulders slope forward • VERY SLOW PROGRESSION
General Dx Tests • Serum Creatinine Phosphokinase (CPK) • Electromyography (EMG) • Muscle Biopsy
General Trx • Supportive • Physical Therapy • Orthopedic Trx (casting, bracing, surgery) to minimize deformities and maintain ability to perform ADL’s
Duchenne • Most severe + most common type • X-linked recessive • Inherited MOTHER carrier/Son Symptoms • Genetic mutation—ABSENT skeletal muscle protein
S/S • Muscle weakness by 3 y/o • Hx delayed motor development • Abnormal Gait, Waddling • Falls Frequently • Marked Lordosis when standing • Gower’s Sign
S/S • Enlarged calves, upper arms, thighs fatty infiltration of muscle pseudohypertrophic • Contractures • 12 y/o = unable to walk • Weakened respiratory muscles • Death
Complications • Contracture Deformities • Atrophy • Trx • PROM & AROM • Casting/Bracing • Rigid Corset • Frequent Rest • PT 3 hrs of ambulaton/day
Complications • Infections d/t decreased vital capacity and atrophy of resp muscles • Obesity d/t overfeeding and decreased activity • Antibiotics • Resp. Trx • Chest Physiotherapy • Diet • Recreation as tol. • Maintain mobility as long as possible
Cardiac Complications • D/T Weakening of Cardiac Muscle • Treatment • Digoxin • Diuretics e.g furosemide
Diagnosis • Serum Enzymes • Creatinine Phosphokinase • Aldoase • Glutamic-oxaloacetic transaminase (SGOT) • Very high levels in 1st 2 years of life • Levels decrease as muscle deteriorates • WNL when severe wasting and disability
Diagnosis • Muscle Biopsy • Degeneration of muscle fibers • Fatty deposits • Fibrosis • EMG • Diminished duration and amplitude of existing motor unit potentials
Nursing Care • Help maintain independence • Continual evaluation of capabilities • Home Assessment • Set-up w/c assessible?, wide doors?, etc • Buying clothes • Respite Care • Family Involvement • Genetic Counseling