Download
1 / 31
310 likes | 583 Vues
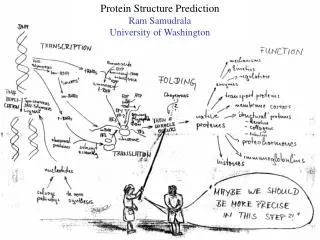
Modelling genome structure and function Ram Samudrala University of Washington. Rationale for understanding protein structure and function. structure determination structure prediction. Protein structure - three dimensional - complicated - mediates function. homology

E N D
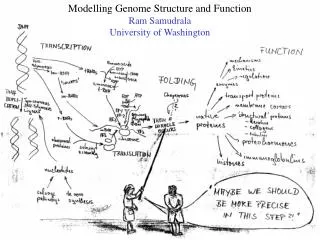
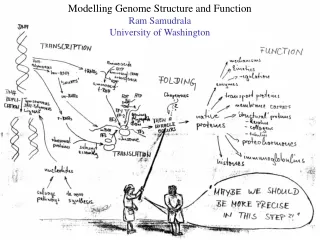
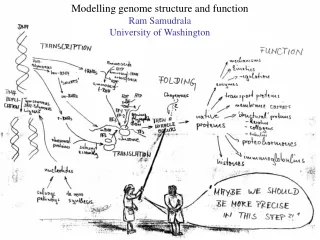
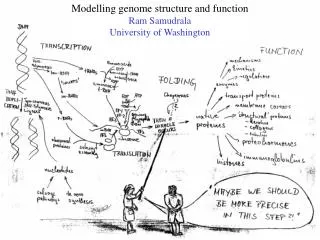
Modelling genome structure and function Ram Samudrala University of Washington
Rationale for understanding protein structure and function structure determination structure prediction Protein structure - three dimensional - complicated - mediates function homology rational mutagenesis biochemical analysis model studies Protein sequence -large numbers of sequences, including whole genomes ? Protein function - rational drug design and treatment of disease - protein and genetic engineering - build networks to model cellular pathways - study organismal function and evolution
Protein folding not unique mobile inactive expanded irregular spontaneous self-organisation (~1 second) native state DNA …-CUA-AAA-GAA-GGU-GUU-AGC-AAG-GUU-… protein sequence …-L-K-E-G-V-S-K-D-… one amino acid unfolded protein
Protein folding not unique mobile inactive expanded irregular spontaneous self-organisation (~1 second) unique shape precisely ordered stable/functional globular/compact helices and sheets native state DNA …-CUA-AAA-GAA-GGU-GUU-AGC-AAG-GUU-… protein sequence …-L-K-E-G-V-S-K-D-… one amino acid unfolded protein
Ab initio prediction of protein structure select sample conformational space such that native-like conformations are found hard to design functions that are not fooled by non-native conformations (“decoys”) astronomically large number of conformations 5 states/100 residues = 5100 = 1070
Semi-exhaustive segment-based folding fragments from database 14-state f,y model generate … … monte carlo with simulated annealing conformational space annealing, GA minimise … … all-atom pairwise interactions, bad contacts compactness, secondary structure filter EFDVILKAAGANKVAVIKAVRGATGLGLKEAKDLVESAPAALKEGVSKDDAEALKKALEEAGAEVEVK
Historical perspective on ab initio prediction Before CASP (BC): “solved” (biased results) CASP2: worse than random with one exception CASP1: worse than random CASP3: consistently predicted correct topology - ~ 6.0 Å for 60+ residues *T56/dnab – 6.8 Å (60 residues; 67-126) **T61/hdea – 7.4 Å (66 residues; 9-74) **T64/sinr – 4.8 Å (68 residues; 1-68) **T75/ets1 – 7.7 Å (77 residues; 55-131) *T74/eps15 – 7.0 Å (60 residues; 154-213) **T59/smd3 – 6.8 Å (46 residues; 30-75) CASP4: ?
Prediction for CASP4 target T110/rbfa Ca RMSD of 4.0 Å for 80 residues (1-80)
Prediction for CASP4 target T97/er29 Ca RMSD of 6.2 Å for 80 residues (18-97)
Prediction for CASP4 target T106/sfrp3 Ca RMSD of 6.2 Å for 70 residues (6-75)
Prediction for CASP4 target T98/sp0a Ca RMSD of 6.0 Å for 60 residues (37-105)
Prediction for CASP4 target T126/omp Ca RMSD of 6.5 Å for 60 residues (87-146)
Prediction for CASP4 target T114/afp1 Ca RMSD of 6.5 Å for 45 residues (36-80)
Postdiction for CASP4 target T102/as48 Ca RMSD of 5.3 Å for 70 residues (1-70)
Historical perspective on ab initio prediction Before CASP (BC): “solved” (biased results) CASP2: worse than random with one exception CASP1: worse than random CASP3: consistently predicted correct topology - ~ 6.0 Å for 60+ residues CASP4: consistently predicted correct topology - ~4-6.0 A for 60-80+ residues *T98/sp0a – 6.0 Å (60 residues; 37-105) **T102/as48 – 5.3 Å (70 residues; 1-70) **T97/er29 – 6.0 Å (80 residues; 18-97) **T106/sfrp3 – 6.2 Å (70 residues; 6-75) **T110/rbfa – 4.0 Å (80 residues; 1-80) *T114/afp1 – 6.5 Å (45 residues; 36-80)
Comparative modelling of protein structure align KDHPFGFAVPTKNPDGTMNLMNWECAIP KDPPAGIGAPQDN----QNIMLWNAVIP ** * * * * * * * ** … … build initial model construct non-conserved side chains and main chains refine
Historical perspective on comparative modelling alignment side chain short loops longer loops BC excellent ~ 80% 1.0 Å 2.0 Å
Historical perspective on comparative modelling alignment side chain short loops longer loops BC excellent ~ 80% 1.0 Å 2.0 Å CASP1 poor ~ 50% ~ 3.0 Å > 5.0 Å
A graph theoretic representation of protein structure -0.6 (V1) represent residues as nodes -0.5 (I) -0.9 (V2) weigh nodes -0.7 (K) -1.0 (F) construct graph -0.6 (V1) -0.2 -0.5 (I) -0.9 (V2) -0.1 -0.5 (I) -0.9 (V2) -0.1 -0.1 -0.3 -0.1 find cliques -0.2 -0.4 -0.3 -0.1 -0.1 -0.4 W = -4.5 -0.2 -0.7 (K) -1.0 (F) -0.2 -0.7 (K) -1.0 (F)
Prediction for CASP4 target T128/sodm Ca RMSD of 1.0 Å for 198 residues (PID 50%)
Prediction for CASP4 target T111/eno Ca RMSD of 1.7 Å for 430 residues (PID 51%)
Prediction for CASP4 target T122/trpa Ca RMSD of 2.9 Å for 241 residues (PID 33%)
Prediction for CASP4 target T125/sp18 Ca RMSD of 4.4 Å for 137 residues (PID 24%)
Prediction for CASP4 target T112/dhso Ca RMSD of 4.9 Å for 348 residues (PID 24%)
Prediction for CASP4 target T92/yeco Ca RMSD of 5.6 Å for 104 residues (PID 12%)
Historical perspective on comparative modelling alignment side chain short loops longer loops BC excellent ~ 80% 1.0 Å 2.0 Å CASP1 poor ~ 50% ~ 3.0 Å > 5.0 Å CASP2 fair ~ 75% ~ 1.0 Å ~ 3.0 Å CASP3 fair ~75% ~ 1.0 Å ~ 2.5 Å CASP4 fair ~75% ~ 1.0 Å ~ 2.0 Å CASP4: overall model accuracy ranging from 1 Å to 6 Å for 50-10% sequence identity **T111/eno – 1.7 Å (430 residues; 51%) **T122/trpa – 2.9 Å (241 residues; 33%) **T128/sodm – 1.0 Å (198 residues; 50%) **T125/sp18 – 4.4 Å (137 residues; 24%) **T112/dhso – 4.9 Å (348 residues; 24%) **T92/yeco – 5.6 Å (104 residues; 12%)
Computational aspects of structural genomics A. sequence space B. comparative modelling C. fold recognition * * * * E. target selection F. analysis D. ab initio prediction * * * * * * * * * * * * * * * * * * * * targets (Figure idea by Steve Brenner.)
Computational aspects of functional genomics structure based methods microenvironment analysis structure comparison sequence based methods sequence comparison motif searches phylogenetic profiles domain fusion analyses zinc binding site? homology function? + assign function to entire protein space * G. assign function * * * * * + experimental data
Conclusions: structure Ab initio prediction can produce low resolution models that may aid gross functional studies Comparative modelling can produce high resolution models that can be used to study detailed function Large scale structure prediction will complement experimental structural genomics efforts
Conclusions:function Detailed analysis of structures can be used to predict protein function, complementing experimental and sequence based techniques Structure comparisons and microenvironment analyses can be used to prediction function on a genome-wide scale Large scale function prediction will complement experimental functional genomics efforts
Take home message Acknowledgements Michael Levitt, Stanford University John Moult, CARB Patrice Koehl, Stanford University Yu Xia, Stanford Univeristy Levitt and Moult groups Prediction of protein structure and function can be used to model whole genomes to understand organismal function and evolution