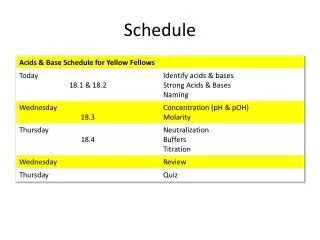
Schedule
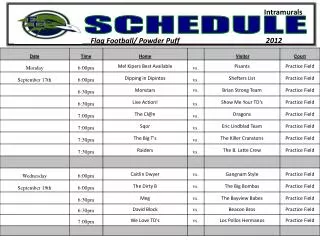
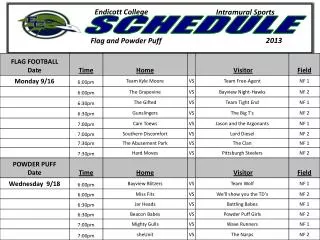
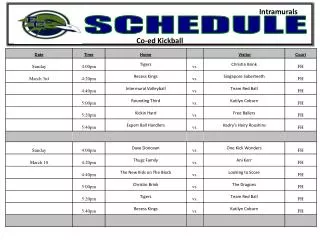
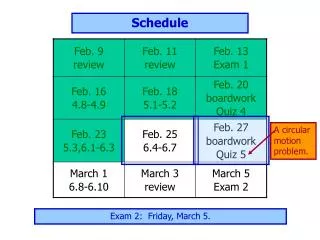
Schedule. Day 1: Molecular Evolution 9.00-10.15 Lecture: Models of Sequence Evolution and Statistical Alignment 10.30-12.00 Practical: Molecular Evolution (Phylogenies – PHYLIP+) 2.00-3.30 Lecture: Molecular Evolution & Comparative Genomics


Schedule
E N D
Presentation Transcript
Schedule Day 1: Molecular Evolution 9.00-10.15 Lecture: Models of Sequence Evolution and Statistical Alignment 10.30-12.00 Practical: Molecular Evolution (Phylogenies – PHYLIP+) 2.00-3.30 Lecture: Molecular Evolution & Comparative Genomics 3.30-5.00 Student Activity: Prepare projects Day 2: Population Biology and Mapping 9.00-10.00 Lecture: Population Genetics and Gene Genealogies 10.00-11.00 Exercise: Jukes-Cantor and Rate Matrix 11.00-12.00 Lecture: Inferring Recombination Histories 2.00-3.30 Practical: DNA Sequence Analysis (PAML Phase +) 3.30-5.00 Student Activity: Prepare projects Day 3: Integrative Genomics (IG) 9.00-10.15 Lecture: High Throughput Data, the structure of IG, GF 10.30-12.00 Practical: Statistical Alignment & Footprinting 2.00-3.30 Lecture (L): Grammars and RNA Prediction 3.30-5.00 Student Activity: Prepare projects Day 4: Integrative Genomics (IG) 9.00-10.00 Lecture: Networks and other concepts 10.00-11.00 Exercise: Stochastic Context Free Grammars 11.00-12.00 Lecture: Concepts, Data Analysis and Functional Studies 2.00-3.30 Practical: Detecting Recombinations 3.30-5.00 Student Activity: Prepare projects Day 5: Project Discussion/Presentation 9.00-10.00 Project 1 – Population Genomics: 1000 genomes 10.00-11.00 Practical – Integrative Data Analysis – Mapping 11.00-12.00 Project 2 – Comparative Genomics: Signals 2.00-3.00 Project 3 – Integrative Genomics: Basic data types 3.00-4.00 Exercise: Statistical Alignment 4.00-5.00 Project 4 – Comparative Biology: Networks
The Data & its growth. 1976/79 The first viral genome –MS2/fX174 1995 The first prokaryotic genome – H. influenzae 1996 The first unicellular eukaryotic genome - Yeast 1997 The first multicellular eukaryotic genome – C.elegans 2000 Arabidopsis thaliana, Drosophila 2001 The human genome 2002 Mouse Genome 2005+ Dog, Marsupial, Rat, Chicken, 12 Drosophilas 1.5.08: Known >10000 viral genomes 2000 prokaryotic genomes 80 Archeobacterial genomes A general increase in data involving higher structures and dynamics of biological systems
The Human Genome (Harding & Sanger) 3*109 bp Myoglobin a globin *50.000 b-globin (chromosome 11) 6*104 bp *20 Exon 3 Exon 1 Exon 2 3*103 bp 5’ flanking 3’ flanking *103 DNA: ATTGCCATGTCGATAATTGGACTATTTGGA 30 bp Protein: aa aa aa aa aa aa aa aa aa aa
ACGCC ACGCC AGGCC AGGCC AGGCT AGGCT AGGGC AGGCT AGGCT AGGTT AGGTT AGTGC Central Problems: History cannot be observed, only end products. ACGTC ACGTC Even if History could be observed, the underlying process couldn’t !!
Some Definitions State space – a set often corresponding of possible observations ie {A,C,G,T}. A random variable, X can take values in the state space with probabilities ie P{X=A} = ¼. The value taken often indicated by small letters - x Stochastic Process is a set of time labeled stochastic variables Xt ie P{X0=A, X1=C, .., X5=G} =.00122 Time can be discrete or continuous, in our context it will almost always mean natural numbers, N {0,1,2,3,4..}, or an interval on the real line, R. Markov Property: ie Time Homogeneity – the process is the same for all t.
Simplifying Assumptions I Probability of Data Biological setup TCGGTA TGGTT a - unknown 1) Only substitutions. s1 TCGGTA s1 TCGGA s2 TGGT-T s2 TGGTT a5 a4 a3 a2 a1 T A T G G G G C T T Data: s1=TCGGTA,s2=TGGTT 2) Processes in different positions of the molecule are independent, so the probability for the whole alignment will be the product of the probabilities of the individual patterns.
Simplifying Assumptions II a l2+l1 l1 = l2 s1i s2i s2i s1i 3) The evolutionary process is the same in all positions 4) Time reversibility: Virtually all models of sequence evolution are time reversible. I.e. πi Pi,j(t) = πj Pj,i(t), where πi is the stationary distribution of i and Pt(i->j) the probability that state i has changed into state j after t time. This implies that
Simplifying assumptions III t1 e A t2 C C 5) The nucleotide at any position evolves following a continuous time Markov Chain. Pi,j(t) continuous time markov chain on the state space {A,C,G,T}. Q - rate matrix: T O A C G T FA -(qA,C+qA,G+qA,T) qA,C qA,G qA,T RC qC,A -(qC,A+qC,G+qC,T) qC, G qC ,T OG qG,A qG,C -(qG,A+qG,C+qG,T) qG,T MT qT,A qT,C qT,G -(qT,A+qT,C+qT,G) 6) The rate matrix, Q, for the continuous time Markov Chain is the same at all times (and often all positions). However, it is possible to let the rate of events, ri, vary from site to site, then the term for passed time, t, will be substituted by ri*t.
Q and P(t) What is the probability of going from i (C?) to j (G?) in time t with rate matrix Q? i. P(0) = I ii. P(e) close to I+eQ for e small iii. P'(0) = Q. iv. lim P(t) has the equilibrium frequencies of the 4 nucleotides in each row v. Waiting time in state j, Tj, P(Tj > t) = eqjjt Expected number of events at equilibrium vi. QE=0Eij=1 (all i,j) vii. PE=E viii. If AB=BA, then eA+B=eAeB.
Jukes-Cantor (JC69): Total Symmetry Rate-matrix, R: T O A C G T F A -3*aa aa R C a -3*aaa O G a a -3* a a M T a a a -3* a Transition prob. after time t, a = a*t: P(equal) = ¼(1 + 3e-4*a ) ~ 1 - 3a P(specific difference) = ¼(1 - e-4*a ) ~ 3a Stationary Distribution: (1,1,1,1)/4.
Principle of Inference: Likelihood Likelihood function L() – the probability of data as function of parameters: L(Q,D) LogLikelihood Function – l(): ln(L(Q,D)) If the data is a series of independent experiments L() will become a product of Likelihoods of each experiment, l() will become the sum of LogLikelihoods of each experiment Likelihood LogLikelihood In Likelihood analysis parameter is not viewed as a random variable.
Exponentiation/Powering of Matrices By eigen values: where If then and Finding L: det (Q-lI)=0 Finding B: (Q-liI)bi=0 JC69: where k ~6-10 Numerically:
Kimura 2-parameter model - K80 TO A C G T F A -2*b-a b a b R Cb -2*b-a b a O Ga b -2*b-a b M Tb a b -2*b-a a = a*t b = b*t Q: start P(t)
Felsenstein81 & Hasegawa, Kishino & Yano 85 Unequal base composition: (Felsenstein, 1981 F81) Qi,j = C*πj i unequal j Rates to frequent nucleotides are high - (π=(πA , πC , πG , πT) T A Tv/Tr = (πT πC +πA πG )/[(πT+πC )(πA+ πG )] G C Tv/Tr & compostion bias (Hasegawa, Kishino & Yano, 1985 HKY85) (a/b)*C*πj i- >j a transition Qi,j = C*πj i- >j a transversion Tv/Tr = (a/b) (πT πC +πA πG )/[(πT+πC )(πA+πG )]
Measuring Selection ThrPro ACGCCA - ThrSer ACGCCG ArgSer AGGCCG - ThrSer ACTCTG AlaSer GCTCTG AlaSer GCACTG ThrSer ACGTCA Certain events have functional consequences and will be selected out. The strength and localization of this selection is of great interest. The selection criteria could in principle be anything, but the selection against amino acid changes is without comparison the most important
i. The Genetic Code 3 classes of sites: 4 2-2 1-1-1-1 4 (3rd) 1-1-1-1 (3rd) ii. TA (2nd) Problems: i. Not all fit into those categories. ii. Change in on site can change the status of another.
Possible events if the genetic code remade from Li,1997 Possible number of substitutions: 61 (codons)*3 (positions)*3 (alternative nucleotides). Substitutions Number Percent Total in all codons 549 100 Synonymous 134 25 Nonsynonymous 415 75 Missense 392 71 Nonsense 23 4
Probabilities: Rates: b start b a a b Kimura’s 2 parameter model & Li’s Model. Selection on the 3 kinds of sites (a,b)(?,?) 1-1-1-1 (f*a,f*b) 2-2 (a,f*b) 4 (a, b)
alpha-globin from rabbit and mouse. Ser Thr Glu Met Cys Leu Met Gly Gly TCA ACT GAG ATG TGT TTA ATG GGG GGA * * * * * * * ** TCG ACA GGG ATA TAT CTA ATG GGT ATA Ser Thr Gly Ile Tyr Leu Met Gly Ile Sites Total Conserved Transitions Transversions 1-1-1-1 274 246 (.8978) 12(.0438) 16(.0584) 2-2 77 51 (.6623) 21(.2727) 5(.0649) 4 78 47 (.6026) 16(.2051) 15(.1923) Z(at,bt) = .50[1+exp(-2at) - 2exp(-t(a+b)] transition Y(at,bt) = .25[1-exp(-2bt )] transversion X(at,bt) = .25[1+exp(-2at) + 2exp(-t(a+b)] identity L(observations,a,b,f)= C(429,274,77,78)* {X(a*f,b*f)246*Y(a*f,b*f)12*Z(a*f,b*f)16}* {X(a,b*f)51*Y(a,b*f)21*Z(a,b*f)5}*{X(a,b)47*Y(a,b)16*Z(a,b)15} where a = at and b = bt. Estimated Parameters: a = 0.3003 b = 0.1871 2*b = 0.3742 (a + 2*b) = 0.6745 f = 0.1663 Transitions Transversions 1-1-1-1 a*f = 0.0500 2*b*f = 0.0622 2-2 a = 0.3004 2*b*f = 0.0622 4 a = 0.3004 2*b = 0.3741 Expected number of: replacement substitutions 35.49 synonymous 75.93 Replacement sites : 246 + (0.3742/0.6744)*77 = 314.72 Silent sites : 429 - 314.72 = 114.28 Ks = .6644 Ka = .1127
Approaches to Sequence Analysis Data {GTCAT,GTTGGT,GTCA,CTCA} Parsimony, similarity, optimisation. GT-CAT GTTGGT GT-CA- CT-CA- Ideal Practice: 1 phase analysis. Actual Practice: 2 phase analysis. statistics s1 s2 s3 s4 TKF91 - The combined substitution/indel process. Acceleration of Basic Algorithm Many Sequence Algorithm MCMC Approaches
T= 0 # - - - ## # # # T = t # # # # s1 r s2 s1 s2 s1 s2 Thorne-Kishino-Felsenstein (1991) Process A # C G * • (birth rate) < m(death rate) 1. P(s) = (1-l/m)(l/m)l pA#A* .. *pT #T l =length(s) 2. Time reversible:
# - - - - - # # # # k * - - - - * # # # # k l & m into Alignment Blocks A. Amino Acids Ignored: # - - - ## # # k e-mt[1-lb](lb)k-1 [1-lb-mb](lb)k [1-lb](lb)k p’k(t) pk(t) p’’k(t) b=[1-e(l-m)t]/[m-le(l-m)t] p’0(t)= mb(t) B. Amino Acids Considered: T - - - RQ S W Pt(T-->R)*pQ*..*pW*p4(t) 4 • T - - - - • R Q S WpR *pQ*..*pW*p’4(t) • 4
Dpk = Dt*[l*(k-1) pk-1 + m*k*pk+1 - (l+m)*k*pk] Dp’k=Dt*[l*(k-1) p’k-1+m*(k+1)*p’k+1-(l+m)*k*p’k+m*pk+1] Dp’’k=Dt*[l*k*p’’k-1+m*(k+1)*p’’k+1- [(k+1)l+km]*p’’k] Differential Equations for p-functions # - - ... - # # # ... # # - - - ... - - # # # ... # * - - - ... - * # # # ... # Initial Conditions: pk(0)= pk’’(0)= p’k (0)= 0 k>1 p1(0)= p0’’(0)= 1. p’0 (0)= 0
Basic Pairwise Recursion (O(length3)) i j Survives: Dies: i-1 i i-1 i j-1 j j i-1 i i j-2 j i-1 j j-1 …………………… …………………… …………………… e-mt[1-lb](lb)k-1, where …………………… …………………… b=[1-e(l-m)t]/[m-le(l-m)t] 0… j (j+1) cases 1… j (j) cases
Basic Pairwise Recursion (O(length3)) survive death j (i-1,j) j-1 (i-1,j-1) Initial condition: p’’=s2[1:j] ………….. (i-1,j-k) ………….. ………….. i-1 i (i,j)
Corner Cutting ~100-1000 Better Numerical Search ~10-100 Ex.: good start guess, 28 evaluations, 3 iterations Accelleration of Pairwise Algorithm (From Hein,Wiuf,Knudsen,Moeller & Wiebling 2000) Simpler Recursion ~3-10 Faster Computers ~250 1991-->2000 ~106
a-globin (141) and b-globin (146) (From Hein,Wiuf,Knudsen,Moeller & Wiebling 2000) 430.108 : -log(a-globin) 327.320 : -log(a-globin -->b-globin) 747.428 : -log(a-globin, b-globin) = -log(l(sumalign)) l*t: 0.0371805 +/- 0.0135899 m*t: 0.0374396 +/- 0.0136846 s*t: 0.91701 +/- 0.119556 E(Length) E(Insertions,Deletions) E(Substitutions) 143.499 5.37255 131.59 Maximum contributing alignment: V-LSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF-DLS--H---GSAQVKGHGKKVADALT VHLTPEEKSAVTALWGKV--NVDEVGGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKVKAHGKKVLGAFS NAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR DGLAHLDNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAHKYH Ratio l(maxalign)/l(sumalign) = 0.00565064
VLSPADNAL.....DLHAHKR 141 AA long *########### …. ### 141 AA long 2 108 years 2 107 years 2 109 years *########### …. ### *########### …. ### ???????????????????? k AA long 109 years The invasion of the immortal link
Algorithm for alignment on star tree (O(length6))(Steel & Hein, 2001) *ACGC *TT GT s2 s1 a s3 *ACG GT *###### * (l/m)
Binary Tree Problem a1a2 * * # # # - - # # # - # TGA ACCT s1 s3 a1 a2 s2 s4 GTT ACG • The ancestral sequences & their alignment was known. ii. The alignment of ancestral alignment columns to leaf sequences was known The problem would be simpler if: How to sum over all possible ancestral sequences and their alignments?: A Markov chain generating ancestral alignments can solve the problem!!
- # # E # # - E * * lb l/m (1- lb)e-m l/m (1- lb)(1- e-m) (1- l/m) (1- lb) # # lb l/m (1- lb)e-m l/m (1- lb)(1- e-m) (1- l/m) (1- lb) _ #lb l/m (1- lb)e-m l/m (1- lb)(1- e-m) (1- l/m) (1- lb) # - lb Generating Ancestral Alignments a1 * a2 * # # l/m (1- lb)e-m E E (1- l/m) (1- lb) - # lb
The Basic Recursion ”Remove 1st step” - recursion: S E ”Remove last step” - recursion: Last/First step removal are inequivalent, but have the same complexities. First step algorithm is the simplest.
Sequence Recursion: First Step Removal Pa(Sk): Epifixes (S[k+1:l]) starting in given MC starts in a. Pa(Sk) = e a E Where P’(kS i,H) = F(kSi,H)
Maximum likelihood phylogeny and alignment Gerton Lunter Istvan Miklos Alexei Drummond Yun Song Human alpha hemoglobin;Human beta hemoglobin; Human myoglobin Bean leghemoglobin Probability of data e-1560.138 Probability of data and alignment e-1593.223 Probability of alignment given data 4.279 * 10-15 = e-33.085 Ratio of insertion-deletions to substitutions: 0.0334
Metropolis-Hastings Statistical Alignment Lunter, Drummond, Miklos, Jensen & Hein, 2005