Download
1 / 11
110 likes | 227 Vues
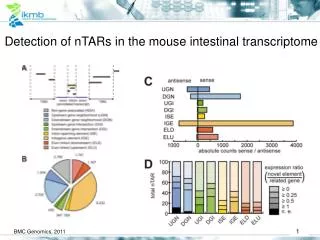
Detection of nTARs in the mouse intestinal transcriptome. BMC Genomics, 2011. nTARs - workflow ( n ovel t ranscriptional a ctive r egion). covered regions. m apping (GEM). yes. yes. yes. discard hit. known exon?. no. discard hit. bad. quality check. no. no. OK.

E N D
Detection of nTARs in the mouse intestinal transcriptome BMC Genomics, 2011
nTARs - workflow(novel transcriptional active region) covered regions mapping (GEM) yes yes yes discard hit known exon? no discard hit bad quality check no no OK linked to known regions/other nTars reading frame (RF) + start codon ? reading frame (RF) ? no discard hit yes new exon? new gene? UTR? check neighborhood check for splice sites
nTARs – raw output position quality • neighborhood/ overlap connections other • chromosome • start • end • avg. mapping quality • avg. base quality • closest genes • + distance • overlapping • known regions • (i.e. introns) • gene • isoform • region type • #supporting pairs • #supporting (splitted) reads • longest RF • (both strands) • earliest start codon • position (both RFs) • sequence around • start- and end point • of splitted reads • (possible splice site)
nTars – example (NA06984.1.M_111124_4.bam) nTar length = 528bp, 120 aa RF at the end nTar avg. base quality = 32.67 nTar avg. mapping quality = 195.61 • short reading frames • no link to known Tar • avg. base quality < 19 for one part (dashed line) • => discard hits link between nTar and RP3-395M20.9 supported by 15 splitted reads / 21 pairs RP3-395M20.7 RP3-395M20.9 CAG|GTGGGG CAG|G
nTar – outlook coverage genome position nTar known exon SNP • identification of exact nTar borders (RF, splitted reads,...) • SNP dependent transcriptional regions • => „eQTL“ for nTARs (1000 Genomes SNPs) • how many nTars overlap with known exons? • find SNP-specific nTars on population seq level • Potential synergy with splice analysis part of main paper....
Subtle splice events Hiller et al. 2006 Permutations of the topic: include GYN(N)nGYN and NAG(N) nNAG with n in our analysis <14
VCF (1000 Genome SNVs) detection of SNVs in proximity of exon ends (i.e. 30 bp) (remember genotypes) BAM list of target ‘splice-sites‘ read Q-filter detection of split-reads overlapping ‘splice-sites’ (each split-read-pattern = potential isoform) generate ‘normalized’ split-read count for all three possible genotypes: ref/ref ref/alt alt/alt Fisher’s exact test for all isoform combinations and genotypes total number of ‘passed filter‘ reads identification of ‘subtle splice’-affecting SNVs
Comparison of every isoform with all possible combinations of the other isoforms isoform 1 isoform 4 isoform 2 isoform 3 example: N = 4 isoforms group 2 2N-2 combinations (= 16-2 = 14) group 1 2(N-1)-1 combinations (= 8-1 = 7) ignore 0000 (all samples in group 2) 1111 (all samples in group 1)
possible combinations for N=4 isoforms black shaded = duplicated combination = 25 unique combinations for 4 isoforms
Consequences of SNVs on tandem splicing predictionphase 1000 Genome SNVs …NAGCGAG CTCGATGTGTGATT… …NAG CGAGCTCGATGTGTGATT… destruction of ‘usually’ realized AG generation of novel AG …NAG CGAXCTCGATGTGAGATT… Do these SNVs actually generate new (or remove) isoforms in individuals carrying the variant?
Population genetics splice-acceptor splice-donor …NAG CGGAGCT GYN… …NAG CGGAGGT GYN… A A AAAAAAA CCCCCCCCC GGGGGGGGG TTTTTTTTT K A G S G A G S T important nucleotide for isoform