Download

1 / 28
350 likes | 1.39k Vues
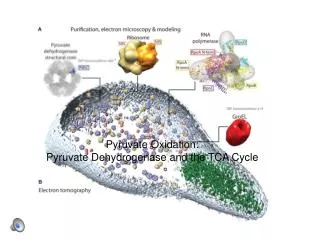
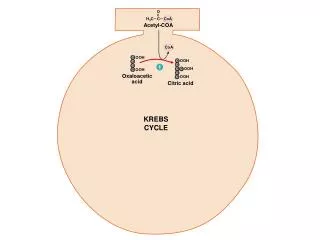
Oxidation of Pyruvate to Acetyl-CoA. Pyruvate formed in the cytosol transported into the mitochondrion Oxidatively decarboxylated to acetyl-CoA NADH produced, transferred to the respiratory chain. 1 mol glucose 2 mol NADH ~ 2 x 2,5 ATP Enzyme : Pyruvate Dehydrogenase complex

E N D
Oxidation of Pyruvate to Acetyl-CoA Pyruvate formed in the cytosol transported into the mitochondrion Oxidatively decarboxylated to acetyl-CoA NADH produced, transferred to the respiratory chain. • 1 mol glucose 2 mol NADH ~ 2 x 2,5 ATP • Enzyme : Pyruvate Dehydrogenase complex • Pyruvate dehydrogenase component inhibited by NADH and Acetyl-CoA. • Deficiency of B1 vitamin beri-beri
Pyruvate is decarboxylated by the pyruvate dehydrogenase component of the enzyme complex to a hydroxyethyl derivative of the thiazole ring of enzyme-bound thiamin diphosphate, which in turn reacts with oxidized lipoamide, the prosthetic group of dihydrolipoyl transacetylase, to form acetyl lipoamide (Figure 18–5). Acetyl lipoamide reacts with coenzyme A to form acetyl-CoA and reduced lipoamide. The reaction is completed when the reduced lipoamide is reoxidized by a flavoprotein, dihydrolipoyl dehydrogenase, containing FAD. Finally, the reduced flavoprotein is oxidized by NAD+ , which in turn transfers reducing equivalents to the respiratory chain. Pyruvate + NAD+ + CoA Acetyl-CoA + NADH + H+ + CO2
Pyruvate Dehydrogenase Is Regulated by End-Product Inhibition & Covalent Modification Pyruvate dehydrogenase is inhibited by its products, acetyl-CoA and NADH. It is also regulated by phosphorylation by a kinase of three serine residues on the pyruvate dehydrogenase component of the multi enzyme complex, resulting in decreased activity and by dephosphorylation by a phosphatase that causes an increase in activity. The kinase is activated by increases in the [ATP]/[ADP], [acetyl-CoA]/[CoA], and [NADH]/[NAD+ ] ratios. Thus, pyruvatedehydrogenase, and therefore glycolysis, is inhibited both when there is adequate ATP available, and also when fatty acids are being oxidized.
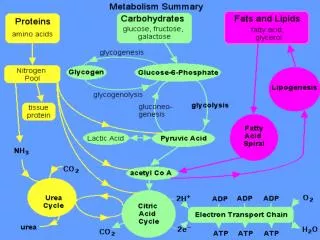
Glycogen: Glycogenesis & glycogenolysis • Energy storage as carbohydrate in the liver and muscle • In the liver up to 6 % w/w • In the muscle up to 4 % w/w • Fasting for 18 hours will depletes glycogen in the liver • In the muscle glycogen will never be depleted • Molecular mass up to 4 millions
Glycogenesis • In the fed state, glycogen synthesis increase • Glucose 6P (some, not all) G 1P (phosphoglucomutase) • G 1P with UTP UDPG + PP (UDPG pyrophosphorylase) • PP Pi drives the reaction to the right (pyrophosphatase) • UDPG + glycogenn glycogenn+1 + UDP (Glycogen Synthase = GS) • UDP + ATP UTP + ADP • Glycogen synthesis from glucose require two ATP
Pathways of glycogenesis and of glycogenolysis in the liver.
Amylo α(1-4) α(1-6) Transglucosidase)
Pathways of glycogenesis and of glycogenolysis in the liver.
Glycogenolysis During fasting(liver) or during contraction (muscle) • Glycogen cleavaged by phosphorylase and debranching enzymes • Glucose 1P ( G 1P ) and glucose are released • Phosphate Pi is required • In the liver : G 1P G 6P (phosphogluco mutase) G 6P G + Pi (glucose 6Pase) • In the muscle : G 1P G 6P (phosphogluco mutase) G 6P enter glycolysis ATP Glucose 6 Pase can only be found in the liver, intestine and kidney
Control of Glycogenolysis During fasting when blood glucose tend to decline, glucagon through protein G will activates adenylyl cyclase. Adenylyl cyclase catalyzes formation of cAMP from ATP. cAMP in turn activates cAMP Dependent Protein Kinase. Then it catalyzes Phosphorylase Kinase into PK-P
Phosphorylase Kinase-P (active = a) will catalyzes Phosphorylase (b=inactive) to Phosphorylase-P (a). The Phosphorylase-P will split Glycogen. Glucose 1P released (require Pi). Glucose 6 phosphatase hydrolysis G 6P to G and Pi. Glucose enter the blood stream to stabilize blood Glucose.
Epinephrine in muscle If you are startled epinephrine in muscle activates Adenylyl cyclase , catalyze ATP cAMP cAMP Dependent PK active. Phosphorylase kinase active. Phosphorylase active. Glycogen G 1P. G 6P no G 6Pase in the muscle, G 6P x G. G 6P glycolysis ATP Eye Central N.S Peripheral nerves Synapses Ca++ Ca/Calmodulin phosphorylase kinase active etc.
Glycogenesis Gycogen Synthase ( GS ) active form GS, and GS-P inactive form. Protein Kinase GS GS-P ↑ GSK There are seven Protein kinase that control glycogenesis Ca++ / Calmodulin Dependent ( 1 phosphorylase kinase ) cAMP Dependent Protein kinase Glycogen Synthase Kinase (GSK) 3, 4 and 5
Glycogenolysis in muscle increases several 100-fold at the onset of contraction; the same signal (increased cytosolic Ca2+ ion concentration) is responsible for initiation of both contraction and glycogenolysis. Muscle phosphorylase kinase, which activates glycogen phosphorylase, is a tetramer of four different subunits, α, β, γ, and δ. The α and βsubunits contain serine residues that are phosphorylated by cAMP-dependent protein kinase. The δ subunit is identical to the Ca2+ -binding protein calmodulin, and binds four Ca2+ . The binding of Ca2+ activates the catalytic site of the subunit even while the enzyme is in the dephosphorylated b state; the phosphorylated a form is only fully activated in the presence of high concentrations of Ca2+ .
Protein Phosphatase-1 Protein Phosphatase-1 hydrolyzes protein-P to Protein Kinase and Pi . Enzymes - P : • Glycogen Synthase-P • Phosphorylase-P • Phosphorylase Kinase-P Protein Phosphatase-1 is inhibited by Inhibitor 1-P Inhibitor 1 Inhibitor 1-P its phosphorylation catalyzed by cAMP Dependent PK.
The Control of Phosphorylase Differs between Liver & Muscle In the Liver: Inhibited by ATP G 6P Glucose In the muscle : Inhibited by: ATP G 6P Activated by 5’AMP
Effect of Insulin: Phosphorylase is inhibited by glucose (in the liver) . Insulin inhibits Phosphorylase if G available. If glucose concentration increases G 6P follows, in turn it will activates glycogen synthase Insulin (+) GS-P GS Protein phosphatase-1
Epinephrine effects liver glycogenolysis • through : • 1.Its effects on glucagon released • 2.Beta-adrenergik receptor, in turn • cAMP formation etc. • 3.Alfa-adrenergic receptor • Inositol triphosphate (IP3), Ca++ • exitfrom ER Phosphorylase kinase • phosphorylase etc. • ( The major mechanism of the three)
Glycogen Storage Diseases Are Inherited Type Ia glycogenosis ( von Gierke's disease ). Deficiency : glucose-6 phosphatase Clinical features : Glycogen accumulation in liver and renal tubule cells, hypoglycemia; lactic acidemia; ketosis; hyperlipemia. Type V (Myophosphorlylase deficiency, McArdle's syndrome) Deficiency : Muscle phosphorylase Poor exercise tolerance; muscle glycogen abnormally high (2.5–4%); blood lactate very low after exercise
Type VI (Hers' disease) Deficiency : Liver phosphorylase Clinical features: Hepatomegaly; accumulation of glycogen in liver; mild hypoglycemia; generally good prognosis Type VII (Tarui's disease) Deficiency : Muscle and erythrocyte phosphofructokinase 1 Clinical features: Poor exercise tolerance; muscle glycogen abnormally high (2.5–4%); blood lactate very low after exercise; also hemolytic anemia