Download

1 / 31
320 likes | 625 Vues
Concepts and methods in genome sequencing and sequence assembly. B. Franz LANG, Département de Biochimie Bureau: H307-15 Courrier électronique : Franz.Lang@Umontreal.ca. Biochemical Methodologies. Purification of intact high molecular weight DNA

E N D
Concepts and methods in genome sequencing and sequence assembly B. Franz LANG, Département de Biochimie Bureau: H307-15 Courrier électronique: Franz.Lang@Umontreal.ca
Biochemical Methodologies • Purification of intact high molecular weight DNA • Cutting or breaking of DNA, purification of fragments within a desired size range • direct sequencing of purified single DNA fragments (PCR amplification of a desired DNA fragment is an option) • cloning of fragments into plasmids or other vectors • Sequencing with various methods: • (Maxam-Gilbert (chemical degradation of purified fragments) • Sanger (in-vitro DNA synthesis using ‘terminators’, use of dideoxi- nucleotides that do not permit chain elongation after their integration • labeling of sequencing reactions, radioactive with P32 or S35, or by fluorescent substances that are detected through specialized optics • Sequencing Artifacts
DNA purification • Cell disintegration • enzymatic (glucanases, cellulases: yeasts, fungi; proteases: animal tissue) • mechanical (glass beads or sand grinding, compression / decompression, sonication etc) • In eukaryotes, possibility of organelle purification (nuclei, mitochondria, chloroplasts) • differential centrifugation (low speed, high speed) • kinetic gradient centrifugation (e.g., sucrose or glycerol) • equilibrium centrifugation (e.g., Percoll)
Gradient centrifugationEquilibrium : organelles (layered on top of a self-forming Percoll gradient) or DNAs/RNAs (separated by self-forming salt gradients, CsCl, KI) migrate to the zone of the gradient that corresponds to the apparent density of the organelle or DNA/RNA molecule. Percoll is made up of polysaccharide beads and gradients form quickly, whereas CsCl or KI gradients require long centrifugation at high g forces. Equilibrium gradients will not change further once they reach a final state.Kinetic (zonal): the molecules or particles migrate from top to bottom (according to the gradient density, the g force, the time, the shape of particles ... etc.). Gradients are usually produced by continuous mixing of sucrose, glycerol, or Percoll solutions of various concentration. Principles of kinetic migration may be combined with equilibrium centrifugation.
DNA Purification continued Other purification procedures • digestion with proteinases, RNases … to remove unwanted contaminants • solvent extractions (phenol, chloroform) • dialysis (removal of low m.w. components like salts and peptides) • CsCl gradient centrifugation (equilibrium) in the presence of DNA-binding fluorochromes like ethidium bromide (BE), DAPI or bisbenzimide (Hoechst dye), which change the apparent DNA density either by • conformation (supercoil/linear: BE) • A+T content (DAPI, Hoechst dye) • agarose gel electrophoresis (large fragments) or polyacrylamide electrophoresis (small fragments) • chromatography (affinity or molecular sieve) • pulsed field electrophoresis (agarose), for chromosome separation or for fragments > 20 kbp
Separation of DNA fragments on gels Agarose/BE PAGE
Cloning of genomic DNA fragments • fragmentation with restriction enzymes • complete digestions (no overlap of fragments) • partial digestions (overlap of fragments but difficult to standardize and requires much starting material) • mechanical fragmentation by ‘nebulisation’ (i.e., passage of DNA solution through a capillary: the viscosity of the DNA solution, the applied pressure and the time of treatment determine the average fragment size. Popular in genome projects. • sonication, or partial DNase digestions (not as random)
Cloning …. The essential part for genome sequencing is here that a single fragment is cloned from one DNA molecule, into a standard site of a vector that flanks the DNA fragment from both sides with known sequence. Cloning allows isolation of large quantities of pure fragment. It further permits the use of pairs of universal primers from which one may sequence into the fragment from both directions (Sanger method of sequencing).
Advantages/disadvantages of different types of vectors for genome sequencing • Phage M13: allows to produce plasmids and single stranded phage from clones; insertion size is small (< 2 kbp). Increasingly less used. • Plasmids: high copy number and easy to purify, insertions limited to < 20 kbp currently the most used vectors • Phage lambda: insertion size up to 20 kbp, efficient cloning (in vitro packaging; positive selection of insertions). Popular for cDNA libraries but difficult to sequence. • Cosmids: derived from lambda, but larger insertions (up to 30 kbp); equally efficient cloning but low DNA yield and difficult to sequence. • BAC (bacterial artificial chromosomes): up to 100 kbp insert and very popular; difficult to sequence directly, but good material for complete random sequencing of the large insert • YAC (yeast artificial chromosomes): chromosome size inserts, however unstable in yeast (recombination). Not (or rarely) used any longer.
Sequencing methods Remember: • DNA is a molecule with a polarity. By convention, sequences are always written from 5’ (left) to 3’ (right), or a different polarity is clearly indicated when this is not the case. • DNA can be cloned and produced as single- or as double-strand (e.g., M13/phagemids versus plasmids)
Sequencing principles Definition according to Maniatis (a popular methods cook-book): The two rapid sequencing techniques in current use are the enzymatic method of Sanger et al. (1977) and the chemical degradation method of Maxam and Gilbert (1977). (Note that Maxam and Gilbert is currently as good as unused, except for special applications such as mapping of protein binding to DNA). Although very different in principle, these two methods both generate separate populations of (radio- or fluorochrome-) labeled oligonucleotides that begin from a fixed residue or combination of residues. … These populations of oligonucleotides are then resolved by electrophoresis under conditions that can discriminate between individual DNAs that differ in length by as little as one nucleotide (PAGE). When the populations are loaded into adjacent lanes of a sequencing gel, the order of nucleotides along the DNA can be read directly from an image (radioactive or optical) of the gel.
Maxam-Gilbert sequencing, chemical modification and cleavage • clonging of DNA fragments, DNA purification (plasmids …) • restriction enzyme digestions, purification • end-labeling of fragments (radioactive, fluorescent) [the following steps lead to DNA fragments that are labeled on only one strand at the same position] • second restriction cut, re-purification of fragments • alternatively, separation of the two DNA strands after denaturation, by migration on partially denaturing PAGE. Single stranded DNAs are extracted after electrophoresis • partial (!) chemical modifications that are specific for one or more nucleotides (see table on next slide) • chemical cleavage with piperidine • dissociation of DNA (!; formamid, heat) and high resolution gel separation (that distinguishes bands differing in one nt length) • autoradiography or fluorescent signal capture with optical system
Maxam-Gilbert sequencing Principles: Purified DNA fragments are labeled
Maxam Gilbert sequencing Labeled DNA 3-5 chemical (partial !)modification reactions chemical cleavage of modified sites
Séquençage Maxam-Gilbert DNA molecules that are ‘visible’ because end-labeled
Maxam-Gilbert - autoradiogram Decoding principle Separation on acrylamide gel of highest resolution; gel concentrations between 3%-20%, for best resolution of different regions (example: 3.5 %)
Maxam-Gilbert sequencing Advantages/disavantages • Requires lots of purified DNA, and many intermediate purification steps • Relatively short readings • Automation not available (sequencers) • Remaining use for ‘footprinting’ (partial protection against DNA modification when proteins bind to specific regions, and that produce ‘holes’ in the sequence ladder) • In contrast, the Sanger sequencing methodology requires little if any DNA purification, no restriction digests, and no labeling of the DNA sequencing template.
Sequencing with dideoxy-nucleotide terminators (Sanger) Termination of DNA (or RNA) synthesis, in the presence of deoxi- plus dideoxi-nucleotides • Cloning and DNA purification (plasmides, M13 …), or purification of total genomic DNA, or PCR amplification … • Dissociation of DNA and hybridization with synthetic primers (may be labeled) • Two primers can be used in the same reaction (example, ‘forward’ and ‘reverse’) if the detection system distinguishes different fluorescent labels (e.g., LiCOR sequencer). In this case, the primers have to be labelled. • Elongation of primer with polymerases (T7, Taq, Thermosequenase …), in the presence of a well-balanced mix of deoxi- and dideoxinucleotides (either primers, nucleotides or terminators may be labeled; MJ, ABI sequencers) • dissociation of DNA (!; formamide, heating) and gel separation (horizontal or vertical; capillaries) • autoradiography (historically), or rather detection of fluorescent signals by sequencer optics
Artifacts in sequencing and sequence assembly The denatured DNA is not linear as it folds back on itself and then migrates differently on the sequencing gel • reason: secondary structures, mainly in G+C –rich regions • effect: ‘compression’ zones in the sequencing ladder • solutions (i) sequence DNA in the two directions of complementary strands; sequencing artifacts due to folding are not symmetric; (ii) for Sanger sequencing, use nucleotide analogs that minimize secondary structure folding, like deaza-NTP, deaza-dITP, or ITP ( instead of NTPs or dGTP, respectively)
Artifacts in sequencing and assembly Sequencing ladders terminate prematurely or contain ‘holes’ Reasons: • sequencing reactions over-modified (M&G), or too elevated terminator concentrations (Sanger); • (ii) strong nucleotide bias, like long runs of A or T that cause many polymerases to fall of the template (Sanger)
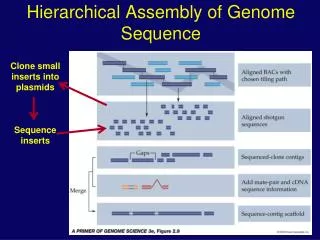
Artifacts in sequencing and assembly Assembly does not progress despite increasing number of readings; contig continue to form and break apart again. Reasons: genomic sequence contains many long stretches of repeats. • direct or inverted clusters of repeats that are longer than the longest single reading which might span this problem zone • long, dispersed genomic repeat elements that are longer than a single reading (like bacterial IS elements of ~ 1 kbp) Solution: sequence plasmids of known inserts size from both sides of insertion (double barrel sequencing strategy)
Available software for sequence assembly Most popular non-commercial software: • ‘Staden package’ (no support since ~ 2000, ‘aging’ applications) • Phred / Phrap / Consed. An assembly of very efficient applications, realtively easy to use (Windows, Mac, Linux/Unix) • Arachne, Tiger assembler ….. (developed by sequencing centers like Broad Institute, JGI …). These applications often require difficult computer installations and are not necessarily available in their most updated form.
Assembly with Phred/Phrap/Consed Phred • reads and re-interprets sequence traces without relying on the interpretation by the sequencer software. All common trace file formats (.abi, .scf …) are automatically recognized. • assigns quality values (probabilities) for every sequence position of a trace, includes recognition of compressions and other artifacts. This requires knowledge about the type of sequencing chemistry, the brand and version of sequencer … documented in a definition file. • removes primer and vector sequences automatically – which requires that these sequences have been previously identified in a definition file. • writes out files (phd) with the inferred sequence and its positional reliability values, up to a user-defined cutoff. The phd files are the material for the assembly, performed by Phrap. The developers provide a script PhredPhrap that integrates the described operations with the assembly, without need of user intervention.
Assembly with Phred/Phrap/Consed Phrap • reads in phd files and performs the assembly of fragments that overlap on one or the other strand, yet accepting mismatches in positions of low confidence, and accepting un-aligned stretches of sequences at the begin (unrecognized short vector sequences or primers) and end of readings. • the algorithm can make use of positional information of readings: forward and reverse readings from the same plasmid or DNA fragment are preferentially positioned close and in opposite direction (‘double barrel sequencing’). • by default, the algorithm attempts a maximum of assembly even in regions of low confidence: assembly errors that are usually eliminated by adding more and better sequence readings. • PhredPhrap does not foresee the elimination of sequence regions of low quality, which have to be recognized by Consed.
Assembly with Phred/Phrap/Consed Consed • reads the assembly made by Phrap, allows to identify potential errors • regions of low cumulative confidence • regions sequenced in only one direction (on only one strand) • positions with contradicting high quality • stretches of single readings that do not match a consensus due to assembly of other readings (polymorphism or assembly errors) • permits writing out of the interpreted final sequence • …. lots of very powerful menu items to be explored