Download

1 / 36
360 likes | 752 Vues
Genome Sequencing and genome viewers. 6/10/ 2014. Lecture outline . Overview of genome sizes and organization Overview of sequencing a genome Next generation sequencing High-throughput experiments by sequencing Genome browsers. Genome sizes. Genome sequencing.

E N D
Genome Sequencing and genome viewers 6/10/2014
Lecture outline • Overview of genome sizes and organization • Overview of sequencing a genome • Next generation sequencing • High-throughput experiments by sequencing • Genome browsers
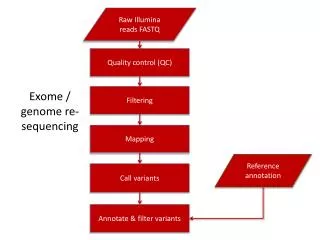
Genome sequencing • Approach depends on the size, complexity and available data for a given organism • De novo sequencing • Re-sequencing for annotation • Sequencing to identify variations • Sample prep – can it be cultured?
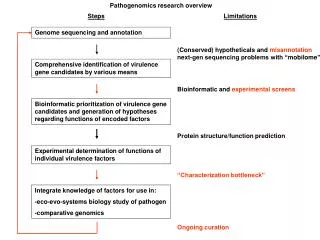
Types of questions • How many genes? • How many functional genetic elements • miRNAs, ncRNAs • What’s different about this genome compared to another one? • Virulence differences in pathogenic organisms • What is the cause of this particular phenotype? • What taxonomic groups are represented in this population of bacteria, viruses or fungi?
Genetic maps • Chromosomal banding patterns • Stain with Giemsa (G-banding pattern) Chromosomes are numbered based on size Giemsa binds to phosphate groups & attaches to regions that are AT rich Dark regions heterchromatic, late replicating and AT rich Lighter regions euchromatic, early replicating and GC rich
Chromosome nomenclature p (petite) = short arm q (queue) = long arm Bands are numbered going away from centromere 4q21.1 represents chromosome 4, long arm 2ndband, 1st sub-band and 1st sub-sub-band
DNA sequencing – Overview • Gel electrophoresis • Predominant in 1980s • Whole genome strategies • Physical mapping (BAC clones) • Walking • Shotgun sequencing • Capillary sequencing machines • Computational fragment assembly • Next generation technologies • Polony based sequencing • Novel assembly techniques Cost/base for DNA sequence
Traditional approach • Shear the very large genome into smaller chunks • Clone in vectors that can support large inserts • Digest and separate on high resolution gel to determine the clone overlap • Pick minimum number of clones • Shotgun sequence each clone • Read the traces and assemble • Make the gene calls • Load it into a genome viewer
Paired reads vs single reads • Single reads • M13 clones • robotic template prep • Paired reads • Plasmids, cosmids, BACs Gap closure!! Prefer 3-10 mate pairs per gap Inserts of different, but known sizes
Steps to Assemble a Genome Some Terminology read a 500-900 long word that comes out of sequencer mate pair a pair of reads from two ends of the same insert fragment contig a contiguous sequence formed by several overlapping reads with no gaps supercontig an ordered and oriented set (scaffold) of contigs, usually by mate pairs consensus sequence derived from the sequence multiple alignment of reads in contig 1. Find overlapping reads 2. Merge some “good” pairs of reads into longer contigs 3. Link contigs to form supercontigs 4. Derive consensus sequence ..ACGATTACAATAGGTT.. Target: 30X coverage or >30 high quality reads per base
Assembled into chromosomes • Refseq nomenclature: • NT: genomic sequence of complete gene • NC: chromosome • NM: mRNA sequence • NP: protein sequence Assembly: completed genome, multiple assemblies
Calling the genes • De novo computer algorithms • Identify coding sequences by GC content • Start and stop sites • Intron/exon boundaries • Comparison with other known genes • EST libraries
Sanger method MishaAngrist
Sanger sequencing reached its technical limits • Only modestly parallel (394 lanes/machine) • Long read lengths (500-900 bp) & >99.9% correct • Need to clone the DNA to obtain enough for sequencing reaction • At SLU: cost for typical Sanger sequencing is $5-6/sample with reliable 500 bp of sequence
NGS sequencing • Polony: discrete clonal amplifications of a single DNA molecule, grown in a gel matrix. The clusters can then be individually sequenced, producing short reads • Polony-based or cluster-based sequencing is the basis of most second generation sequencers Typical NGS workflow: Library construction to add adapters to sequence Template CLONAL amplification (on a bead or chip) Massively PARALLEL sequencing
IlluminaHiSeq and miSeq • 100 – 200 bp read lengths • Available locally with MoGene and Cofactor Genomics • GTAC (Wash U) has HiSeq 2000 which has 50bp single end reads and 100 bp paired-end reads • Why not use this for all sequencing? • Cost is ~300-400/library and ~$1100/lane of sequencing • Generate Gb of data per run
Ion Torrent – measures pH changes Done on a semi-conductor chip
Illuminavs Ion Torrent • Illumina has greater capacity but longer run times • Latest versions of both have read lengths ~200 bp • SLU will soon have an Ion Torrent machine • Costs?? • Can do single- or pair-end reads • Paired end are 2X cost for library construction, but necessary for de novo genome assembly
Bioinformatics challenges • Each flow cell in the IlluminaHiseq 2000 can generate a billion bases of sequence • Raw read files are Tb in size • Processed read files are several 700-800 Mb • Alignment files 150-300 Mb • Assembly of millions of short (75-100 bp) reads into vertebrate genome • Need high-performance compute (HPC) cluster for vertebrate sized genomes
Sequencing has become a standard technique • RNA sequencing for expression • ChIP sequencing for TF site identification • DNA sequencing for variants • Identification of populations/genetic changes in highly variable viruses and bacteria • Metagenomics • Identification of unknown/non-culturable communities of bacteria/viruses/fungi
Why RNAseq over microarray? • Technical variation is less • Do not need a sequenced genome • Greater dynamic range of expression • Detect transcript isoforms • Identify novel transcripts • Identify non-coding RNAs
Genome viewers • Provide genomic context for genes • View different tracks of information • Variants, alternative transcripts, GC rich regions • Export images of the gene models • Export genomic sequence for specific regions • Both browsers (UCSC Genome and Ensembl) provide interface for complex queries
Configure tracks Ensembl viewer UCSC viewer
Issues with genome browsers • Different data depending on assembly • Sometimes older assemblies have more data tracks • The rate at which data gets added (and verified) with a new assembly depends on staffing/funding and priorities. • In the case of C. elegans, the Ensembl browser is more up-to-date • For the purpose of our exercises, you can use whatever browser you choose, just be sure to note which one & which assembly you used. • I may recommend one over the other because of certain tool availability
This week in lab • Tutorials on using UCSC and Ensembl genome browsers • Comparing gene structures using genome browsers • Exporting variation data from genomic regions using genome browsers • Designing virtual gene knock-outs